Abstract
Addictions are heritable and unfold dynamically across the lifespan. One prominent neurobiological theory proposes that substance-induced changes in neural circuitry promote the progression of addiction. Genome-wide association studies have begun to characterize the polygenic architecture undergirding addiction liability and revealed that genetic loci associated with risk can be divided into those associated with a general broad spectrum liability to addiction and those associated with drug-specific addiction risk. In this Perspective, we integrate these genomic findings with our current understanding of the neurobiology of addiction to propose a new genetically informed neurobiology of addiction (GINA) model.
Introduction
The loss of life and socioeconomic costs associated with addictive substances burden the world. There are vast individual differences in patterns of substance use, which range from casual or occasional to excessive and disordered. Addiction emerges when, following chronic regular use, the presence of a substance helps maintain homeostasis. Behaviorally and psychologically addiction typically results in the attenuation of reward elicited by initial substance use and the development of compulsive use [G] to ameliorate negative affect as well as psychological and physiological stress states and withdrawal symptoms that arise when a substance is absent. Increasing quantities, dosage, and potencies of substances are often pursued in addiction in an attempt to obtain the increasingly fleeting ‘highs’ experienced during initial use. Box 1 provides an outline of the correspondence between this definition of addiction with the definitions of substance use and substance use disorders (SUDs).
Translational neuroscience research has transformed our understanding of addiction and led to its re-characterization as a neurobiological state rather than a controllable moral failing. A largely independent line of research has shown that the moderate-to-large heritability [G] of SUDs is undergirded by a polygenic [G] architecture that is associated with broad spectrum liability to addiction as well as distinct genetic architectures [G] associated with substance-specific risks. This genetic risk is compounded by environmental factors that are substance-specific (such as policy-related access to specific substances) and associated with factors related to general psychiatric liability (such as socioeconomic status).
In this Perspective, we incorporate knowledge from human genetic studies of addiction into brain disease models. First, we provide an overview of the three-stage neurobiological model of addiction, which postulates that substance-induced neural changes are the predominant contributor to the etiology of SUDs. We then showcase how contemporary genetic work has begun to identify the polygenic architecture underlying addiction risk. Here, we highlight the utility of genetically-informed study designs to probe the plausibility of proposed models of addiction. Last, we integrate genetic research into an expanded version of the stage-based neurobiological model of addiction by proposing a new model: the genetically informed neurobiology of addiction (GINA) model. In this model, the interplay between genetic liability [G] and substance-induced changes in the neural substrates of positive reinforcement [G] (thought to drive binging [G], intoxication and escalating use), negative urgency [G] (thought to take place during withdrawal [G] and negative affect) and executive function [G] and/or regulatory capacity (important for the preoccupation with and/or withdrawal from substance use), as well as neural and peripheral substance-specific pathways, contribute to SUD development.
Brain-based models of addiction
According to the most prominent neurobiological model of addiction, substance and/or experience-dependent alterations in cortiostriatal and corticolimbic circuitry (Fig. 1) drive three recurring and non-mutually exclusive stages of addiction: binge-intoxication, withdrawal-negative affect and preoccupation-anticipation.
Fig 1: Corticostriatal and corticolimbic circuits underlying addiction.
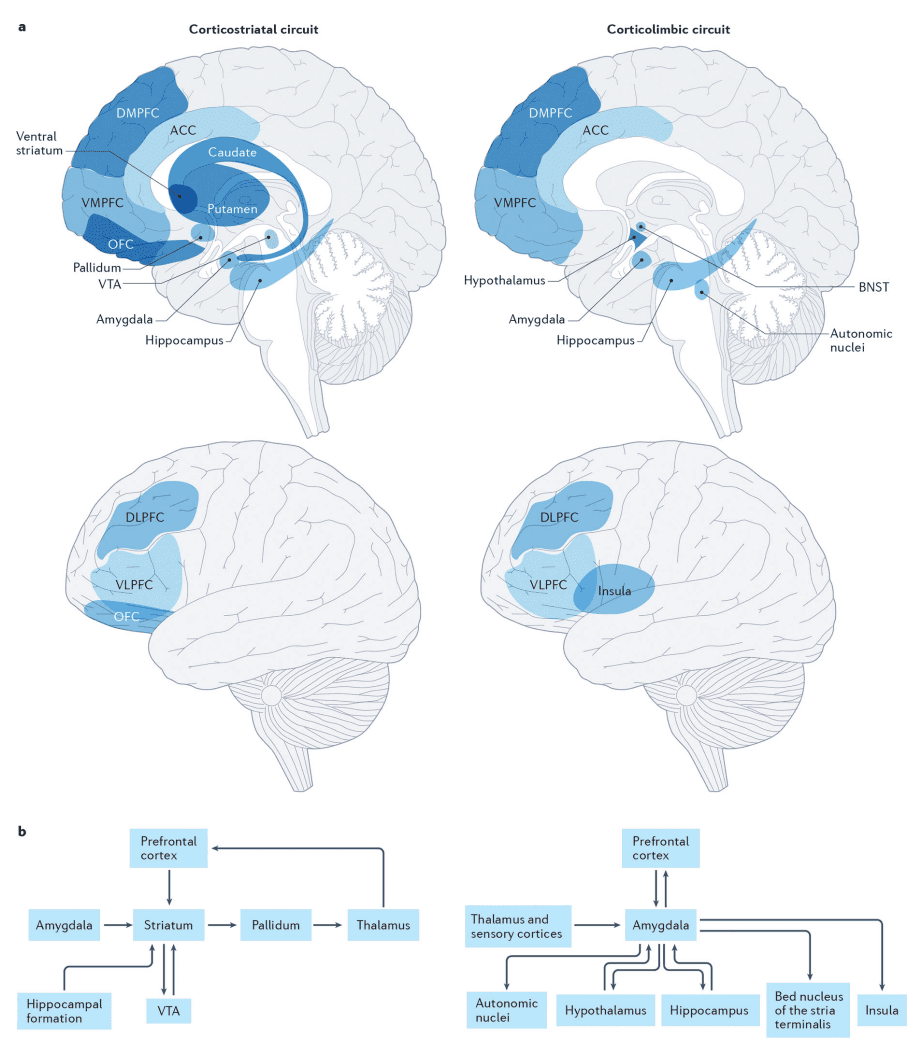
Anatomical locations of (a) and connections between (b) the primary nodes within the corticostriatal and corticolimbic circuits that support reward, emotion, and their regulation and are proposed to influence the binge-intoxication, withdrawal-negative affect, and preoccupation-anticipation stages of addiction. The corticostriatal circuit is critical for reward processing and largely contributes to the binge-intoxication stage of addiction. The striatum (comprised of the putamen, caudate and ventral striatum) is the primary node of this network. Through its connections with other nodes, the striatum supports learning reward contingencies, hedonic responsiveness, generating motivation to pursue rewards and goals, forming and implementing plans to obtain reward, adjusting behavior and plans according to changing contingencies, and coordinating motor movements in the service of obtaining reward. More specifically, dopaminergic projections from the ventral tegmental area to the nucleus accumbens within the ventral striatum support reward prediction and learning in combination with multimodal sensory information received from the basolateral amygdala and contextual information from the hippocampal formation. Projections from the striatum to the pallidum support hedonic responsiveness through endogenous opioid stimulation and provide motivational signals to the VMPFC (supporting integration of contextual and interoceptive information, bottom-up drive and top-down regulation) and the DLPFC (supporting goal-directed planning) through thalamic relays. Afferents from the PFC to the ventral striatum further serve to facilitate the implementation of plans to obtain reward (DLPFC) as well as flexible behavioral adjustment when expected actions do not obtain predicted outcomes (ACC) and can facilitate or inhibit the motivational significance of reward predictive cues in the environment. The corticolimbic circuit is critical for affective processing and behavioral vigilance; it largely contributes to the withdrawal-negative affect stage of addiction. The amygdala (inclusive of the amygdala and the extended amygdala) is the primary node of this network; through its connections with other nodes it supports responses to environmental challenges, including threat and stress, by generating and regulating emotional responses. Low and high resolution sensory information arrives in the basolateral complex of the amygdala from the thalamus and sensory cortices, respectively. Efferent projections from the centromedial and extended amygdala, including the bed nucleus of the stria terminalis (BNST), to autonomic nuclei (such as the parabrachial nucleus), the hypothalamus and the hippocampus drive emotional responses, including fear conditioning and the generation of stress-related physiological changes. Direct and indirect connections between the amygdala and insula facilitate interoception (awareness and importance of our physiological states). Projections from the nucleus basalis of Meynert in the extended amygdala facilitate amygdala-driven arousal and sensitivity of the cortex. Projections from the amygdala to the VMPFC promote subjective awareness and evaluation of emotion and the integration of affective information (such as motivational information conveyed by the ventral striatum projections shown in the upper panel). Projections from the DLPFC and VLPFC to the amygdala through the DMPFC and VMPFC promote the regulation of affective responses and physiological arousal. Both the corticostriatal and corticolimbic circuits support executive function and the regulation of behavior to influence the preoccupation-anticipation stage of addiction by contributing to incentive salience (such as the ventral striatum projections to the VMPFC and OFC within the corticostriatal circuit), interoceptive signals associated with withdrawal physiology and affect (such as the insula within the corticolimbic circuit), as well as the regulation of behavior (through the DLPFC, VLPFC and ACC in both circuits). While there are many additional connections within and between these circuits, we present a heuristic model focusing on those most well linked to addiction. These circuits are explained in greater detail in prior publications. Note: Unlike prior depictions of the stage-based neurobiological model which show 3 circuits corresponding to each stage, we present the corticostriatal and corticolimbic circuits, which are hypothesized to predominantly drive the binge-intoxication and withdrawal-negative affect stages, respectively. The preoccupation-anticipation stage is undergirded by prefrontal connections within and across these circuits in this model.
During the binge-intoxication stage of the neurobiological model of addiction, substance-induced stimulation of neural reward circuitry provides positive reinforcement. In support of this idea, all addictive substances have been shown to directly or indirectly elicit fast and large increases in dopamine (DA) release in the nucleus accumbens (NAc) that resemble predictive reward signals [G]. Extrinsic cues (such as with whom, where and when one uses a substance) and intrinsic cues (such as mood and physiology) are quickly and strongly paired to drug-reward and become conditioned predictors that motivate substance use. This stage exerts its largest influence on addiction during the initial escalating and episodic heavy use of a substance (particularly during adolescence and young adulthood), the re-emergent escalating use that follows abstinence, and the use of substance types with increasing psychoactive impact (higher doses or potency) following addiction progression.
With continued heavy substance use and progression towards severe SUD, it is proposed that the reinforcing properties of substances shift to negative reinforcement [G] . This transition to the withdrawal-negative affect stage of addiction is marked by distress and anhedonia [G], as well as by the aversive physiological states (such as blackouts, nausea and insomnia) and psychological states (such as anxiety, depression and heightened stress) that arise in the absence of a drug’s effects. During this stage, substance use is compulsive and functions to provide relief from these aversive states by returning the body to a state of homeostasis. Such homeostasis can now only be achieved when the drug is present, because a series of neuroadaptations (such as fewer DA receptors and attenuated reward-related DA release) have taken place as natural adjustments to repeated drug exposure. These adaptations also promote anhedonia, through which other non-drug rewards (such as social interactions, achievement, food and sex) lose their reinforcing properties. Thus, increasing quantities of the addictive substance are required to reach homeostasis, with even greater amounts being required to ‘chase the highs’ that were associated with the binge-intoxication stage. The chronic use of addictive substances also promotes stress and negative affect (through, for example, elevations of corticotropin-releasing hormone in the extended amygdala). During substance abstinence, withdrawal and related negative affect leads to heightened interoceptive salience, through which the physiological arousal associated with withdrawal and negative emotionality potentiates negative reinforcement-related craving [G].
In the third stage of this model, the repeated pairing of drug use with reward and relief results in a cognitive preoccupation-anticipation of the drug in expectation of these effects. This is often characterized by the subjective experience of drug craving. Building on dual systems models that postulate that self-control arises when deliberative executive function counteracts more automatic emotionally driven behavior, this stage of addiction is proposed to be defined by a loss of this regulatory capacity as a result of substance-induced impairment of top-down executive function (such as reduced prefrontal control over striatal and other limbic circuits). Thus, it is proposed that heavy and sustained use causes the prefrontal cortex to become less efficient at minimizing the direct incentive salience [G] evoked by substance cues and emergent negative emotionality, leaving individuals with less intrinsic ability to combat the throes of addiction, even if there are deep subjective desires to stop.
This model emphasizes how substance-induced neural changes contribute to the development and maintenance of moderate to severe SUDs. Given that drugs of abuse impact neurotransmitters and neuromodulators at a magnitude that does not intrinsically occur, it might be assumed that resulting brain changes would be so penetrant that addiction would be a foregone conclusion in anyone with escalating levels of use. In reality, only some individuals exposed to addictive substances develop addiction and a significant minority of individuals with SUDs recover. The neurobiological stage-based model of addiction therefore acknowledges that factors that influence an individual’s predisposition to addiction must be considered as major contributors to the disorder.
The neurobiological stage-based model provides a framework of addiction susceptibility that is not substance-specific. While the concept of an addiction risk that is shared across substances is supported by evidence that SUDs are frequently comorbid with one another and share similar neural, genetic and environmental correlates, emerging evidence reported in a recent preprint highlights substance-specific risk factors that take the form of variants in genes within substance-specific metabolic and signaling pathways. Thus, we can hypothesize that predispositional liability to the addictive properties of a specific substance (or a non-substance-related behavior) may set the pace for transitions between the neurobiological stages of addiction, and intensify the subjective experience of each stage.
Developmental vulnerability [G] and trait-like vulnerability [G] have been highlighted by other neurobiological theories of addiction. For instance, arising from developmental psychology, the neurodevelopmental model of addiction has theorized that adolescence and young adulthood confer broad spectrum addiction risk owing to typical patterns of brain maturation that initially prioritize emotional and social processing over cognitive control and regulation. This promotes risk-taking behavior as well as increased impulsive attempts to cope with negative emotion, placing adolescents and young adults at risk for both the positive and negative reinforcing aspects of SUD risk (particularly in the context of underdeveloped physiological tolerance and still-developing regulatory capacity). In this model, the typical earlier maturation of reward-related and stress and negative affect-related neural circuitry and relatively delayed prefrontal development are seen as addiction risk factors. It is speculated that substance use may more profoundly shape neural circuitry, and especially prefrontal development, during these periods of extensive neural maturation.
The neurodevelopmental and stage-based neurobiological models of addiction have unique origins and differentially weight predispositional liability [G] and substance-induced alterations. The neurobiological model arose primarily from data on functional differences in brain activity and receptor densities and emphasizes the role of substance-induced neural plasticity in the etiology of addiction. On the other hand, the neurodevelopmental model relies predominantly on emergent structural changes during adolescence and emphasizes predispositional developmental liability. However, both models highlight the contributions of impulsivity, negative affect, and executive function (and their neural substrates) in addiction vulnerability.
Over the past five years, we have witnessed immense progress in human genetics (reviewed below) that can shed further light on the mechanisms of addiction. With this in mind, it is now important to begin to measure and integrate predispositional genetic risk into brain disease models of addiction.
Genetics of addiction
Genetics of substance use and SUDs
Historically, the search for genetic variation underlying SUDs has focused on the genes encoding substance-specific neurotransmitters or metabolic enzymes (such as opioid or nicotinic receptors, alcohol dehydrogenase and cytochrome p450) and genes encoding proteins involved in canonical systems that have a widespread effect on psychiatrically relevant behaviors (such as dopaminergic and serotonergic receptors and transporters). As in studies of other complex phenotypes, studies were conducted on relatively modestly sized samples and single gene variants [G], genes, or haplotypes were examined, with exonic variation prioritized. However, the introduction of genome-wide association studies (GWASs) [G] to the field led to a transition from candidate gene [G] validation to genetic exploration, with some unexpected consequences.
For many psychiatric disorders (such as schizophrenia or major depression), the candidate genes that had been hypothesized to be involved in disease liability were found by GWASs to be no more likely to be associated with disease risk than those selected at random and, with a few exceptions, novel loci were associated with these disorders. However, in the case of substance use and addiction, some of the strongest significant signals (in addition to novel loci) in GWASs were in candidate genes known to regulate metabolism (such as ADH1B for alcohol and CYP2A6 for nicotine), encode receptor binding sites (such as CHRNA5 for nicotine and OPRM1 for opioids) and implicated in addiction models (such as corticotropin-releasing factor receptor 1 (CRHR1)). Recent comprehensive reviews of these GWAS findings are available (Box 1).
More generally, GWASs of complex traits (including substance use and SUDs) have revealed that, with a few exceptions (such as the effects of the single nucleotide polymorphism (SNP) [G] rs1229984 in ADH1B on alcohol use), single genetic variants have small effect sizes and that these traits are highly polygenic. Notably, while twin studies [G] suggest a heritability of ~50% for a range of addictions, GWASs and whole exome analyses can explain, at best, only a quarter of this heritability, although the inclusion of less common variants does improve the heritability of some traits. This discrepancy is typical of most complex traits; it is possible that some of this ‘missing heritability’ resides in rare variants that will be identified using sequencing technologies. However, the high polygenicity of addictions also suggest that larger sample sizes may be required to identify additional novel common variants than is the case for other complex traits.
Three other insights unique to SUDs are notable. First, while genetic correlations between liability to substance use (for example likelihood of ever using or frequency of use) and problematic or disordered use are moderate to high, the genetics of disordered use faithfully reproduce a pattern of correlated medical comorbidities — both psychiatric and somatic — and potential indicators of negative life outcomes (such as lower education attainment) whereas the genetics of substance use has been related to adaptive psychosocial correlates and inconclusively linked with psychopathology (Box 1). Accordingly, the risk of substance use may represent a mixture of risk for future problems and resilience to them.
A second insight recapitulates prior evidence from twin studies that genetic liability for SUDs is largely shared across substances, but that there is also important substance-specific liability (Fig. 2). A recent preprint reports that the polygenic architecture underlying the general liability to SUDs includes loci that regulate dopaminergic signaling, including signals linked to the D2 dopamine receptor (DRD2) and cAMP-specific 3’,5’-cyclic phosphodiesterase 4B (PDE4B). As PDE4B is instrumental in neuroplasticity within prefrontal dopaminergic pathways and is associated with stress and negative affect in animal models it represents an intriguing locus for addiction that is consistent with the stage-based neurobiological model of addiction. Another recent GWAS identified genes contributing to shared liability to substance use, addictions and other related behaviors (such as a large number of sexual partners). Genes identified as being associated with traits with externalizing features included cell adhesion molecule 2 (CADM2), which has also been linked to general liability to SUDs as well as to substance use, risky sexual behavior, self-control and obesity. It is thus possible that CADM2 impacts addiction liability by influencing early risk-taking and self-control more broadly (as opposed to influencing addiction progression). Intriguingly, despite the identification of overlapping loci and a high genetic correlation between the externalizing GWAS and a factor identified in the recently-reported GWAS representing common addiction liability, many novel loci are associated with the latter addiction factor, implying that addiction pathology is partially genetically distinct from general liability to externalizing behaviors.
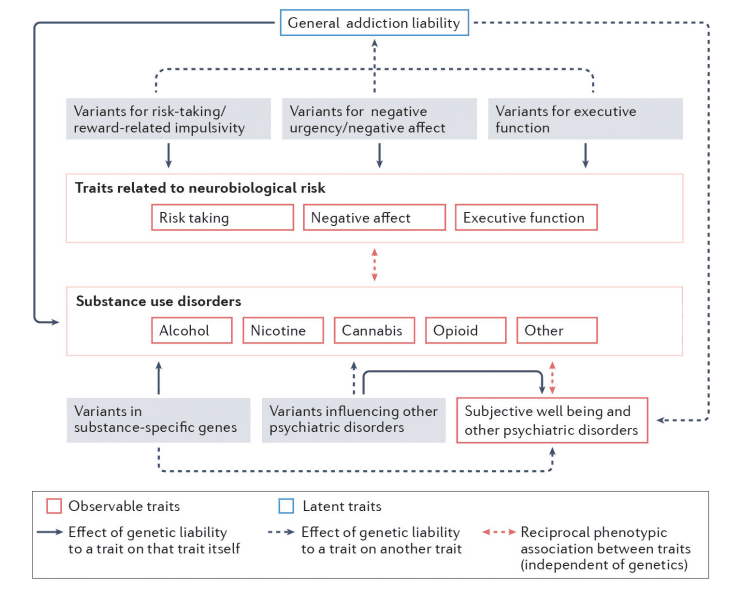
The genetic contribution to individual substance use disorders (SUDs) is attributable to variants that influence general addiction liability and substance-specific variants. General addiction liability is driven by variants influencing traits that correspond to the 3 stages of the neurobiological model of addiction: reward and risk-taking, negative affect and urgency, and executive functioning. In contrast, variants in receptors that respond to the psychoactive components of individual substances or those in genes metabolizing individual drugs directly influence each substance use disorder in a substance-specific manner. Furthermore, genetic variants that influence other psychiatric disorders may also independently influence SUDs (solid arrows indicate the effects of variants on a specific trait/phenotype, whereas the dashed lines indicate cross-trait effects). Reciprocally, the genetics underlying general addiction liability may impact other psychiatric disorder risk (gray dashed arrow). Small effects of substance-specific genetic variants on other psychiatric disordersare also predicted (gray dashed arrow). In addition to these genetic pathways, prolonged substance use and SUDs may phenotypically influence risk-taking, negative affect, executive functioning, as well as psychiatric health and well-being (double headed dashed red arrows depict phenotypic associations). Note that alcohol, nicotine, cannabis, cocaine and opioid use disorders are shown, as there are current large genome-wide association studies of these SUDs; however, the genetics of many other SUDs could be similarly classified.
Third and finally, these GWASs have shown that, after the common genetic liability to addiction is taken into account, residual and substance-specific variation is often conferred by variants in genes encoding metabolic factors and substance-specific receptors. This substance-specific variation is also polygenic; however the effect sizes of some individual variants are an order of magnitude larger than those of the variants that are common across addictions.
Correspondence between GWAS and molecular genetics
That addictions originate and induce perturbations in brain-based genetic pathways is supported by sources of genetic data in addition to GWASs. Many loci linked to addiction through GWASs have been shown to be expression quantitative trait loci (eQTL) [G] across developmental stages in tissue obtained from brain regions implicated in the three-stage neurobiological model of addiction. For instance, variants linked to substance use were associated with gene expression and co-expression network modules in the NAc, anterior cingulate cortex (ACC) (Fig. 1), cerebellum, dorsolateral prefrontal cortex and other brain regions. Gene co-expression patterns were preserved across these brain regions, supporting a generalized overall enrichment of these variants in the brain rather than in specific brain regions. Beyond eQTL effects, heritability enrichment analyses of GWASs have revealed that the genetics of substance use and addiction liability is enriched for tissue- and cell-specific regulatory elements [G] specifically related to chromatin architecture (that is, DNA folding) in primary brain regions specified in the 3-stage neurobiological model of addiction. Genes linked to alcohol and tobacco use and problematic use also show higher expression in excitatory neurons in cortical and midbrain regions, as well as the hippocampus, thalamus and amygdala. Thus, addiction-relevant GWAS have begun to reveal evidence that is convergent with the 3-stage neurobiological model of addiction and highlights the role of predisposition within this framework.
Drug exposure-induced transcriptomic changes have also been observed. In one study, human alcohol dependence GWAS-associated genes showed networks of co-expression in the prefrontal cortex, NAc and ventral tegmental area of ethanol-exposed mouse brains. Another found cross-species conservation in gene expression changes associated with cocaine exposure within the hippocampus and ventral tegmental area that mapped to well-established addiction pathways (such as dopaminergic networks). These studies suggest that, just as the brain responds dynamically to repeated drug exposure, so does the transcriptome and epigenome. However, this genomic and neural plasticity may not be a consequence of drug exposure alone: novel studies that integrate GWAS signals with multi-omics data have suggested that a subset of the genes that are differentially expressed also show enriched genetic associations with substance use. In one study, genes that were differentially expressed in the dorsolateral prefrontal cortex (DLPFC) of individuals that were alcohol-dependent coalesced into two co-expression modules that included genes that were enriched in alcohol use and addiction GWASs. In other instances, genes identified as having significant association with substance use in GWASs were not differentially expressed, but genes within their co-expression networks were. Therefore, GWAS data in combination with multi-omics and cross-species findings can provide insights into the neural gene networks vulnerable to substance-induced modulation.
Brain imaging genetics
The conceptualization of brain structure and function as intermediate phenotypes, or endophenotypes (which are hypothesized to lie between genes and/or environmental experiences and disease processes) generated enthusiasm that genetic research on neural phenotypes would help characterize genetic architecture underlying complex behavior, including addiction. For example, the high heritability of brain structure alongside its objective quantification, reliability and proximity to gene function were the basis for presuming that GWASs of brain structure would yield loci with large effects. Meta-analyses of the GWASs of structural brain phenotypes found, instead, that brain imaging phenotypes are highly polygenic and complex and that their genetic correlation with behavioral outcomes is far more modest than hypothesized. For example, initial genetic correlations (rG) estimated across GWASs of substance involvement (including use, problematic use and SUDs) and brain structure have revealed that, much like the genetic correlations between behavioral phenotypes and substance use phenotypes, the genetic correlations between substance use and/ or SUDs and brain structure are modest in magnitude.. These data suggest that, while there is gene–brain–behavior genetic overlap, it will be difficult to characterize without large samples. Notably, these genetic correlations may be constrained by small effects of brain–behavior associations.
Unraveling the genetic architecture of functional neuroimaging phenotypes (such as task-related activity or resting-state functional connectivity [G]) has been even more challenging. GWASs of task-related functional magnetic resonance imaging (t-fMRI) and resting state functional connectivity phenotypes in the UK Biobank have revealed low heritability and identified few loci. This may be partially attributable to the low reliability of traditional t-fMRI and the need for large amounts of resting state functional connectivity data to facilitate its reliable measurement. Thus, despite the prominence of functional neuroimaging studies of addiction, their vanishingly low heritability, psychometric challenges and practical difficulties in harmonizing the findings of different studies have led to relatively few well-powered genetically-informed investigations
Predispositional and/or Causal?
As reviewed above, GWASs and transcriptomic studies of addiction-related phenotypes highlight the role of predisposition within the 3-stage neurobiological model as well as potential substance-induced changes in epigenetic structure and the transcriptomic landscape. By contrast, the vast majority of brain-imaging addiction-related science has interpreted cross-sectional associations between substance involvement and brain phenotypes to putatively reflect causative substance-induced brain alterations. Below we review evidence from longitudinal and genetically-informed designs that can be used to infer whether substance-related variability in brain structure may plausibly reflect predispositional risk and/or sequela of substance involvement. Much like GWAS, these data highlight the need to incorporate genetic predisposition into neurobiological models of addiction.
Longitudinal studies.
Longitudinal studies of substance involvement have revealed that changes in brain structure and function are associated with escalating substance use. For example, the National Consortium on Alcohol and Neurodevelopment in Adolescence (NCANDA) has found that heavy alcohol use in adolescence is associated with, and precedes, accelerated cortical gray matter decline, particularly in the medial and dorsal prefrontal cortices, as well as a decline in white matter integrity. Similarly, in a cohort examined as part of the IMAGEN study, the initiation of cannabis use was associated with cortical thinning in the superior and anterior medial prefrontal cortex. This change occurred over a 5 year period among participants who were cannabis naïve at baseline (age 14) and in a dose dependent manner that was also associated with impulsivity. Other longitudinal studies, however, have found evidence supportive of the predispositional model. For instance, reduced DLPFC volume in substance-naïve children was associated with an earlier age of drinking initiation when those children reached adolescence and an attenuation of the typical reduction in drinking that occurs in young adulthood. Similarly, two studies found that lower orbitofrontal cortex (OFC) volume in early adolescence preceded and was associated with a subsequent onset of cannabis use.
Two considerations are noteworthy when interpreting longitudinal studies. First, adolescence is characterized by dynamic changes in brain development. As such, individual differences in the trajectories of brain development in youth who initiate substance use may reflect substance-induced changes and/or gradations of predispositional influences on neurodevelopment. While experimental evidence in non-human animals shows that heavy substance use can reduce markers of neurogenesis and brain growth, other evidence suggests that neural development is significantly genetic in origin. Therefore, disentangling the aspects of brain development that are attributable to genetic predisposition from those that are sequelae of substance use is challenging, especially when the genes contributing to brain development may exert pleiotropic effects [G] on substance involvement.
Second, while a dose-response relationship (in which the more one uses a substance, the stronger the association with brain metrics) may reflect causal effects, it is also possible that those with preexisting neurobiological liabilities that manifest in altered neurodevelopmental trajectories in adolescence may be more vulnerable to escalating and disordered substance use. Indeed, GWASs show that increasing severity of drug use is associated with a polygenic signal that is partially distinct from the genes influencing lighter or milder use and is more likely to exert pleiotropic effects on brain development (even when we make simplified assumptions that the effects are linear).
Genetic causal modeling.
Genomic data can be used to examine the plausibility of hypotheses of phenotypic causality. Because modest but significant genetic correlations have been demonstrated between psychiatric phenotypes and brain phenotypes, the associations between variants at addiction-relevant loci and neural phenotypes have been partially attributed to the pleiotropic effects of these variants. However, if addiction is conceptualized as the escalation of drug exposure and the brain phenotype as the target of such prolonged exposure, then any effect of variants in drug-related loci on the brain may represent genetic causality (and this could be tested via Mendelian Randomization; Fig. 3). Modern genetic causality approaches that account for the polygenic nature of SUDs can estimate the proportion of shared genetic liability or identify genetic variants that might be causal. One such analysis found no support for a genetically causal effect of problematic alcohol use on brain structure phenotypes. By contrast, and consistent with a predispositional model, it did provide support for the idea that differences in brain structure (including a lower volume of the basal forebrain and a greater volume of the pars opercularis) may plausibly contribute to problematic alcohol use.
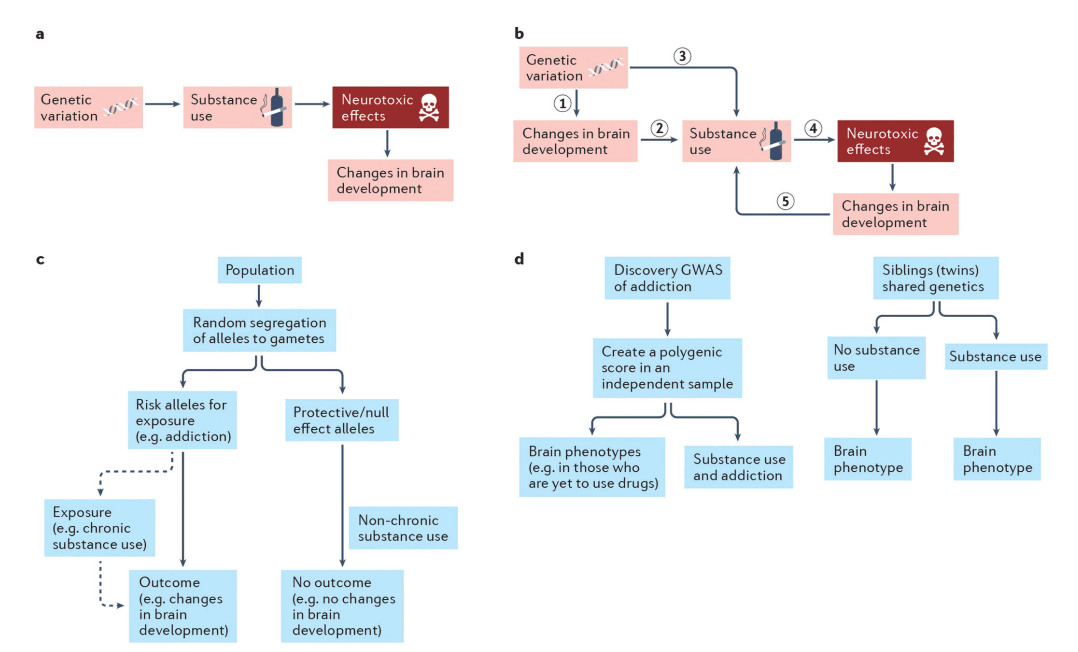
a| According to neurobiological models of addiction, genetic variation influences substance use, which may, in turn, exert neurotoxic effects that alter brain development. b| According to predispositional models of addiction, genetic risk for substance use disorders impacts brain development (1) prior to or concurrent with the onset of and escalating substance use and sets the neurobiological stage for substance use and future addiction (2). Consequent substance involvement (3) (also influenced by genetic risk that is not associated with neural phenotypes) may then causally influence the brain, via neurotoxic mechanisms, to further potentiate problematic substance use (4). Cyclically, these brain-related changes may further enhance risk for addiction progression (5). c| Mendelian randomization and other genetic causal methods can be used to evaluate these models. These approaches are based on the fact that parental genotypes conferring risk of exposure (i.e., chronic substance use) are equally as likely to be inherited by the offspring as genotypes that are protective or of no effect. Individuals inheriting risk alleles or polygenic risk of substance use will subsequently be more likely to use drugs; we can then test whether this chronic use causally alters brain development. In this method, the individual risk alleles or the polygenic risk of drug exposure is the genetic instrument and an independent association between this genetic instrument and the outcome (changes in brain development), as shown in the flow chart, is possible evidence for causal effects of substance exposure on the brain. The genetic instrument is assumed to influence the outcome (changes in brain development) solely via its influence on chronic substance use (dashed line). For a greater discussion of Mendelian Randomization approaches as well as their limitations see. d| Testing the association between polygenic risk for addiction and brain imaging phenotypes, including trajectories, in drug-naïve individuals (left flow chart) is an ideal approach to assess whether pre-existing brain-related differences precede addiction. Here, the effects of genetic variants are taken from a discovery GWAS of addiction and applied to a sample, ideally of individuals without a history of substance use (e.g., children), which has brain data. A polygenic score is created in this new independent sample. It is expected that this polygenic score will eventually be associated with substance use and addiction in this sample. However, if it is also associated with brain phenotypes prior to use of substances, then we can infer that genetic risk that precedes onset of substance use contributes to brain development (part b, step 1) and later substance use (part b, step.3), rather than a causal effect of substance use on the brain alone (part b, step.4). Alternatively, examining twins (or similarly aged non-twin siblings) that are discordant for substance involvement can provide information on whether substance-related neural phenotypes arise from predispositional influences and/or are induced through substance involvement (right flow chart). If the brains of genetically similar individuals differ as a function of their substance use, then non-genetic mechanisms, including substance-induced changes, might be implicated. However, if they brain phenotypes are similar among those discordant for substance use, this would suggest that predispositional effects including shared genetic variation and environmental exposures are responsible for their associations with substance involvement.
Discordant Designs.
Monozygotic (identical) twins that are discordant for drug exposure serve as a natural quasi-experiment for the study of causal effects of drugs in humans (Fig. 3). If the correlation between drug use and brain structure is entirely genetic, the brain structure in monozygotic pairs discordant for substance use (or SUDs) would not be different. If, on the other hand, there is a difference in brain structure, then contributors beyond shared genetics and prenatal and familial environment are implicated. These contributors may be exposure-specific (that is, causal effects) or due to a person-specific third variables (such as early trauma that motivates substance use and, independently, modifies brain structure in that twin). While it is ideal to study identical twins, dizygotic twins and even non-twin siblings (close in age) can also be used to further parse non-genetic sources of similarity (for example, dizygotic twins are more closely matched for prenatal exposures than non-twin siblings).
Several studies have examined structural brain differences in twin pairs who vary in their drug exposure. In data from the Human Connectome Project, it was shown that an association between cannabis use and reduced volume of subcortical structures were no longer apparent when cannabis-using individuals were compared with their co-twins or age-approximate siblings, consistent with the robust estimates of genetic correlation between cannabis use and subcortical brain volume. Similarly, it was discovered that the correlations between alcohol consumption and insula and DLPFC volumes were primarily attributable to predispositional factors. Instead of relying on discordancy for alcohol use alone, this study also contrasted brain structure in twin pairs in which both individuals were heavy drinkers as well as in pairs in which both individuals were light drinkers. Heavy and light drinking twins from discordant pairs did not differ in their gray matter volume and also did not differ from twins in pairs where both were heavy drinkers, suggesting that the reduced gray matter volume that is associated with alcohol use does not arise as a consequence of use. A recent series of studies of 436 24-year old twins from the Minnesota Twin Family Study document support for both predispositional and causal contributors to associations between substance involvement and cortical thickness. For instance, a thinner medial OFC among those with alcohol, tobacco and/or cannabis use disorders was attributable to predispositional risk, with some evidence that alcohol or cannabis use disorder may also contribute to these reductions. In the same sample, an index of alcohol (but not cannabis) use was associated with a thinner cortex overall, with evidence that this reflects both predispositional risk and a potential consequence of alcohol exposure. Collectively, these analyses reveal that brain structure correlates of substance use and SUDs may reflect a predispositional liability to substance involvement, as well as a potential consequence of exposure.
Genetic predisposition.
Evidence for correlations between genetic variants identified in GWASs of brain imaging and those identified in GWASs of substance use and SUDs suggest that associations between brain structure and addiction-related behavior that occur after the onset of exposure are confounded by preexisting pleiotropic liability. Thus, nearly any evidence for causation would also support predisposition. Ideal support for predispositional effects arises from studies of brain development in individuals before their drug exposure. Family studies provide persuasive support for such pre-existing brain differences in those genetically enriched for addiction liability. Youth with a family history of alcohol use disorder have thinner frontal and parietal cortices and smaller frontal gray matter volume, as well as larger gray matter volumes of the cerebellar lobes, the NAc and the amygdala, and task-related response variability in brain regions related to reward response and decision-making than those without such a family history. These studies provide compelling support for the association between family history of addiction and brain structure and function, which (in some instances) was investigated prior to substance use onset. They further support the neurodevelopmental hypothesis because family history of alcohol use disorder was also associated with early behavioral undercontrol.
However, family history is an amalgamation of inherited genetic risk and genetic nurture[G] and can be biased by lack of adequate measurement of familial density of risk. Twin studies disentangle these familial effects to some degree by either explicitly modeling genetic nurture or separating genetic and family environmental factors within the offspring. Well-powered neuroimaging studies that also assess family history and future substance use, particularly during the developmental period prior to onset of substance use, are rare; however, the Adolescent Brain and Cognitive Development (ABCD study) provides an opportunity to evaluate the interrelationships between brain and substance use development in individuals from ages 9–10 years into early adulthood. In the baseline data from this sample, total brain and regional cortical and subcortical volumes, cortical thickness and surface area, fractional anisotropy [G] and mean diffusivity indices were examined for their association with polygenic liability to alcohol consumption and problem drinking (Fig. 3). The polygenic risk score (PRS, the aggregated effects of risk alleles associated with a trait) for problematic alcohol use was associated with lower volume of the left frontal pole and greater cortical thickness of the right supramarginal gyrus, although nominally significant associations for both typical and problematic alcohol use PRS and insula metrics were evident. In another analysis, the PRS for cannabis use disorder, but not cannabis use, was associated with lower white matter volume. In each of these analyses, data were excluded from the small subset of youths who report substance use. However, an even earlier epoch of substance exposure — prenatal exposure — also merits consideration. In the ABCD sample, prenatal exposure to cannabis, particularly beyond the first trimester, is correlated with psychopathology (but not global brain structure) outcomes and persists as children enter their teens. Therefore, the study of predisposition that is exclusively related to genotype requires consideration of family history, genetic nurture and other third variable confounders, as well as prenatal exposure to substance use. However, even studies of prenatal exposure are confounded by intergenerational transmission of genetic predisposition.
The GINA Model
Brain imaging studies of addiction have tended to invoke drug-induced mechanisms of effect, whereas genomic studies have mostly relied on predispositional aspects of vulnerability. Each domain implicitly considers the relevance of the other to some degree. For instance, the neurodevelopmental model references common latent genetic influences on behavioral undercontrol and substance use. Similarly, genomic studies are identifying drug-induced epigenetic alterations in relevant brain regions.
Based on these foundational discoveries we outline an integrated framework for the development of addiction, the Genetically Informed Neurobiology of Addiction (GINA) model (Fig. 4). At the core of the GINA model is polygenic liability. The neural circuitry underlying reward, negative affect and executive function, as well as drug-specific pathways (such as those featuring drug receptors or enzymes involved in drug metabolism) serve as the substrates within which polygenic liability, risk and sequela of addiction unfold. Environmental factors serve as the filter through which gene–brain associations influence addiction-related behavior. While the GINA model is presented as a framework for integrating imaging and genetics studies of addiction in both clinical and population cohorts, it can also be extended to evaluate onset of use, occasional use and milder forms of SUD.
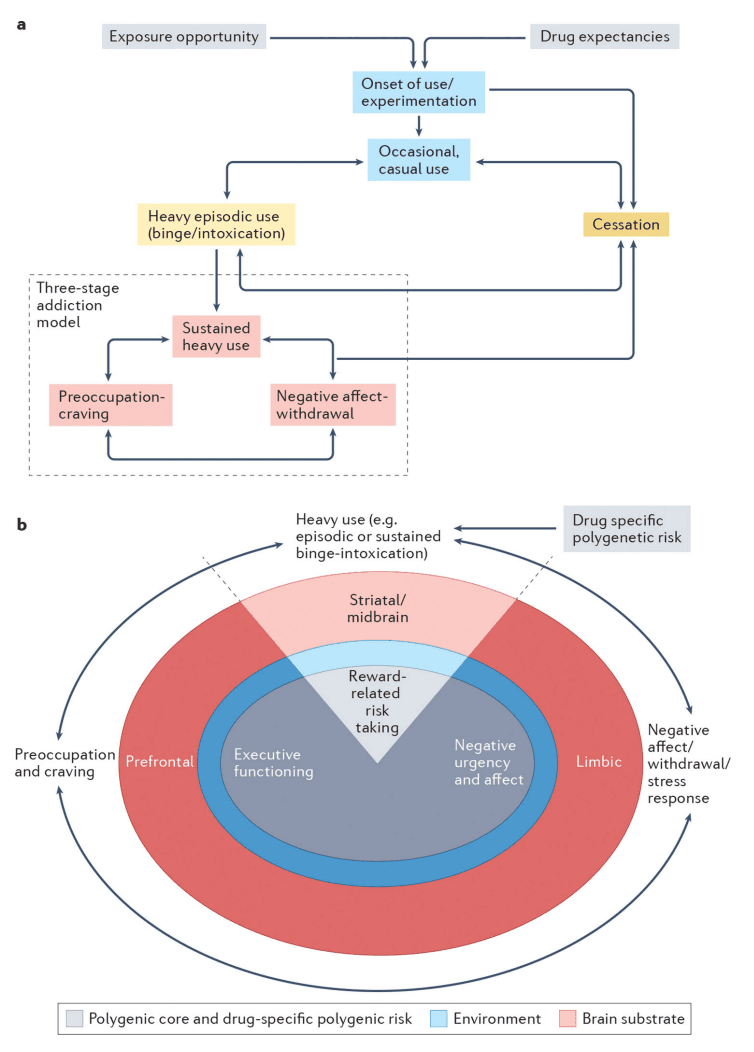
Addiction may be conceptualized as a developmental process or as a syndrome comprised of stages of escalating problem use. While the GINA model described here outlines a testable gene–brain–behavior mechanism underpinning the stages of addiction, it is scalable and can be extended to advance our understanding of the process of addiction. a| An illustration of the process of addiction, and those that lead into addiction, serves as a framework for understanding the GINA model. Exposure opportunity, availability and initial expectations surrounding the anticipated subjective effects of substance use serve as early contributors to drug-seeking behaviors and increase the likelihood of substance use. Onset of substance use occurs in a subset of individuals, with some further entering a phase of casual but repeated substance use. Depending on the addictive potential of the substance, progression through periods of heavy episodic use and cessation may then occur (intervening aspects of these processes are not depicted). For some substances, periods of primarily reward-related occasional or casual use, heavy episodic use and cessation may occur (e.g., heavy drinking limited to college), during which time individuals may even meet criteria for milder forms of SUD. Not shown are the numerous genetic and environmental influences that promote or deter progression through these substance-interfacing behaviors. For a further subset of individuals, heavy episodic use advances into a phase of sustained heavy use, wherein the pleasurable aspects of substance use are attenuated and compulsive use emerges to ameliorate negative affect, psychological and/or physiological stress states, and physiological withdrawal symptoms. Withdrawal, and related negative mood, following substance abstinence leads to potentiated interoceptive salience through which physiological arousal associated with withdrawal and negative emotionality are potentiated. We propose that this phase reflects moderate to severe forms of SUDs. b| The neurobiological model of addiction in the GINA framework. The GINA model places the 3 stages of addiction (shown around the outside of the image) within the context of a polygenic core (grey), environmental filter (blue) and brain substrate (pink; width of circles does not correspond to any relative magnitude of effect, i.e., genes and environment may be equally important). Each stage aligns most closely with genetic predispositions that act via specific posited brain mechanisms (these are shown “stacked” below that stage of the addiction cycle). All of the addiction stages are influenced by a polygenic core, which broadly corresponds to trait representations of substance-induced stages of sustained heavy use (binge-intoxication), negative affect (withdrawal-negative affect), and preoccupation/craving (preoccupation-anticipation) and by additional drug-specific polygenic risk that influences addiction, partly via the brain (for example, variants in genes encoding neurotransmitters) as well as via non-brain mechanisms (such as metabolic variants). Polygenic liability to reward-related risk-taking contributes to initial phases of binge-intoxication and may promote later escalating use (shown as heavy - episodic and sustained - use), which plays a role in promoting the reward related neural response to pleasurable aspects of substance use (e.g., striatal brain regions). On the other hand, chronic substance use induces brain-related alterations that culminate in heightened stress states and negative affect (for instance, those with polygenic liability to negative urgency may be more vulnerable to this pathway) via a modified limbic response. Furthermore, polygenic liability to executive function is likely to be instrumental in drug craving via changes in prefrontal brain function that results in increasing difficulties regulating the emotional salience of substance-related stimuli, despite the potential of subjective and cognitive desires to stop. Despite the appearance in this schematic of a one-to-one correspondence between polygenic liability, brain region and addiction stage, the gene–brain–behavior map is likely to be more interconnected. For instance, sustained heavy use in the context of negative affect may be influenced by polygenic risk to negative urgency and affect, via limbic pathways as well as substance-induced alterations in striatal circuits. The environment provides a filter for genetic liability (i.e., the magnitude and nature of genetic effects may be different in differing environmental contexts) and also directly underpins addictions. The brain is depicted as the outer substrate from which psychological aspects of addiction emerge. While distinct brain systems are illustrated, it is likely that networks of brain regions correspond to the three stages of addiction. While not noted here, aspects of addiction arise from and impact other bodily systems as well as the brain.
Polygenic core
As a simplifying principle, we posit that four key domains of genetic risk form the polygenic core of addictions. Three are common to all addictions: genes affecting reward and risk-taking (notably in the context of positive urgency [G]), genes affecting negative affect and/or susceptibility to negative urgency and genes affecting executive functioning and/or regulation. Genetic measurement of these domains, especially as they pertain to addiction liability, remains incomplete and underspecified. For instance, negative urgency is a hallmark characteristic of SUDs and some comorbid mood disorders. However, current GWASs of negative affect rely on heterogeneous constructs (such as depression or neuroticism). The genetic disarticulation of the sub-facets of these composites (such as negative urgency) as well as well-powered GWASs of addiction-relevant indices of negative affect (such as using substances to cope or stress-responsivity), using approaches such as genomic structural equation modeling [G] will be required to fine-tune this polygenic core from an index of generalized risk for psychopathology to an addiction-specific liability factor.
The fourth source of genetic variability arises from genes encoding drug-specific metabolic factors and receptors; while polygenic in architecture, some of the drug-specific single loci may exert relatively large effects. For instance, the effect sizes of variants in alcohol metabolizing enzymes can approach those seen for the apolipoprotein e4 variant and Alzheimer disease risk. Studying such pronounced (but scarce) genetic effects alongside polygenic patterns of common and drug-specific genetic risk requires novel statistical approaches that can handle mixtures of distributions of genetic effect sizes and conditional analyses. Drug-specific loci that encode the neurotransmitter targets for a drug are, however, rarely so specific. For instance, the rs16969968 variant in CHRNA5, encoding a nicotinic receptor, was shown to be highly significant for tobacco use phenotypes and also associated with schizophrenia and educational attainment. Neuroimaging studies of this variant (rs16969968) have linked the risk allele to greater hippocampal activation in response to smoking cues and with reduced resting-state connectivity between the dorsal ACC and the ventral-striatopallidal circuit but with null effects on brain differences in light smoking adolescents. Drug-specific loci also capture some variability in responses to existing treatments for addiction but findings are mixed and specific GWASs of pharmacogenomics response are needed. Notably, drug-specific loci are evident in GWASs of both substance use and addiction (for example, the rs1229984 variant in ADH1B is significant for typical, maximum habitual and problem and/or disordered drinking), suggesting that their influence on addiction may be routed via their regulation of substance consumption and the subjective, and possibly interoceptive, effects associated with use. Therefore, in the GINA model, we place drug-specific polygenic liability in the context of exposure, and most notably, heavy episodic and heavy sustained use (Fig. 4), where it regulates subjective response and sets the pace for entry into, and progression within, the three-stage addiction model.
Brain substrate
The GINA model provides a framework for gene–brain–addiction mechanisms from which we can develop testable hypotheses. For example, polygenic liability to executive function deficits might modify prefrontal regulatory capacity and, in turn, potentiate preoccupation with drugs. It would, however, be reductive to assume a one-to-one correspondence between polygenic liability, brain region and behavioral manifestation. For instance, it is highly likely that striatal circuitry is sensitive to the stages of addiction that evoke positive urgency and that polygenic liability to risk-taking as well as executive function (that is, undercontrol) undergird positive urgency. Polygenic liability to risk-taking is also likely to contribute to other stages of substance use and addiction (such as relapse) and to affect other brain regions beyond striatal regions (Fig. 4). Multi-method studies that integrate polygenic risk with whole brain and multivariate behavioral phenotypes will be necessary to broaden the scope of gene–brain–addiction connections. For example, machine learning [G] based approaches coupled with large-scale data could be used to perform a systematic, data driven study of the complexity underlying the GINA model.
Environmental filter.
While not detailed in this Perspective, the GINA model incorporates environment as the filter through which addiction emerges. Similar to genetics, some environmental factors (such as life stress) will generalize across substances and other psychopathology while others (such as policy, taxation and distance to alcohol outlet) are likely to be more substance-specific (although policies do have cross-cutting effects). A proportion of the environmental impact on addiction also involves neurobiological mechanisms. For instance, trauma (especially when occurring during early life) is associated with brain development and exacerbates addiction risk. However, the environment and genetic susceptibility may be related. Some traumatic experiences are correlates of genetic risk (such as passive exposure to early adverse environments that are a product of parental genotype), while others are modifiers of polygenic liability (such as trauma that moderates polygenic liability to addiction) and gene expression (such as trauma-induced epigenetic changes).
Characterizing addiction in the GINA model.
The GINA model characterizes addiction as a transition from episodic to sustained heavy use in the context of emerging negative affect and preoccupation (Fig. 4). From a diagnostic perspective, this coincides with moderate and severe SUD, as defined in DSM-5. Recently, individuals endorsing between 2 to 5 DSM-5 criteria (mild or moderate SUD) were classified as being in a high-risk, sub-threshold state of preaddiction (similar to pre-diabetes) where interventions may be maximally beneficial. While the GINA model is more closely aligned with addiction per se, those with the higher preaddiction scores, depending on their individual symptomatology, may well be described by the GINA model. While reminiscent of the three-stage neurobiological model, the GINA model separates the broader binge-intoxication stage into heavy use that is either episodic or sustained. The former represents intermittent reward-motivated accelerations in use (such as alcohol consumption in college students) and may also capture mild SUDs as defined in DSM-5 and thus, is somewhat distinct from addiction. The latter represents the form of escalating chronic use that aligns with current conceptualizations of heavy use in the context of addiction. While behaviorally distinct, these aspects of binge-intoxication may share genetic and neurobiological contributors.
Characterizing substance use in the GINA model.
It could be argued that, rather than being classified as disorders in their own right, addictions would be better represented as a process or as a series of interactions with psychoactive substances that – in some instances – becomes disordered. From this perspective, the addiction process (Fig. 4) begins with exposure opportunity and expectations regarding the drug use experience. These early stages are strongly motivated by environmental factors, although gene–environment correlations influence drug availability and exposure opportunity. Upon onset, initial experiences (which may be heritable) and subsequent experiences (which may be subjective) with individual drugs motivate or deter further use. Continued use represents a mixture of pathways – heavy episodic use may become entrenched and transition to addiction or, for substances with lower addiction potential, settle into patterns of socially accepted or intermittent use. Many individuals attempt to quit using drugs in their 20s and 30s and the GINA model features both this early and later cessation. The same genetic core that contributes to the stage-based neurobiological model is also likely to contribute to these aspects of the addiction process. For instance, genetic propensity to risk-taking may motivate early drug-seeking behaviors and some neuroimaging studies suggest pre-existing brain differences in youth at risk for substance use onset. Likewise, initial and typical subjective responses to individual substances may be influenced by variation in drug-specific loci.
Relationship with other heuristics
The GINA model represents our conceptualization of the vast complexities that underlie addictions and is inspired by the extensive output of psychology, psychiatry, neuroscience, genetics, and translational research generated by international teams of scientists, particularly those who forecasted a need to bridge brain and genome research. It is certainly not unique in adopting a multi-factorial view. Instead, it represents a conceptual increment that has resulted from novel study designs, genetic discoveries and rapid increases in availability of genetically informed neuroimaging data. For instance, the Addictions Neuroclinical Assessment (ANA) was developed to guide researchers in designing studies that might test the three-stage neurobiological model of addiction. However, the GINA model provides a framework from which a series of hypotheses can be tested using the ANA-derived data. A previous study also integrated evidence from behavior genetic and neuroimaging studies to provide a framework for a common liability to addictions and tested it using multiple sources of data. These studies were prescient in anticipating the role of dopaminergic pathways on common addiction risk, although the GINA model has the advantage of leveraging contemporary insights from GWAS to advance a polygenic framework. Other schema, such as the Hierarchical Taxonomy of Psychopathology (HiTOP), aim to outline the common genetic and neurobiological underpinnings of a broad range of psychopathologies, including addictions. While the GINA model includes cross-disorder components (Fig. 2), it is clear that addictions are not merely the product of generalized genetic liability to broad-spectrum psychopathology and chronic substance use. In spirit, the GINA model is aligned with the newly hypothesized Etiologic, Theory-Based, Ontogenetic Hierarchical Framework of Alcohol Use Disorder (ETOH) which highlights the importance of distinguishing between common and drug-specific mechanisms of risk and considering premorbid and drug-acquired pathways of influence. However, the ETOH model is finely tailored towards advancing our understanding of alcohol use disorder, whereas the GINA model focuses on addictions more broadly in the context of common and drug-specific genetic liability.
Predisposition in the context of drug-induced change.
The central aim of the GINA model is to establish the role of genetic predisposition as a source of individual differences in substance-related neural phenotypes. Most neuroimaging studies speculate neurobiological associations arise as sequelae of drug use, while widespread evidence of pleiotropy motivates geneticists to be partial to correlational mechanisms. The GINA model presents one possible reconciliation of these disparate perspectives. Simply put, while the consequence of chronic drug use may be evident in the brain, genetic factors that predate onset of substance use may influence whether or not an individual will initiate drug use, escalate in use, and progress to addiction. Furthermore, trait correlates of substance-induced states serve as additional sources of individual differences (Fig. 4). For example, we theorize that limbic adaptation to chronic drug use is likely to induce a negative affect state in most individuals, but that those with a genetic predisposition to negative affect may be more susceptible to this transition. Thus, individual differences in the developmental tempo and severity of the individual stages are likely to be due to polygenic liability – either via direct effects of genomic variation or via early influences on brain variability – similar to environmental moderators of risk.
Conclusions and perspectives
Future directions
Heuristic models, such as the GINA model, are intended to be dynamic and riddled with chasms that anticipated advances in genetics, neuroscience, and psychiatry can bridge. Recent criticism of genetic approaches surrounds the questionable prognostic utility of addiction PRSs. Neuroimaging data are also associated with small effects and researchers in this field have grappled with their own methodological challenges, such as the reliability and heritability of task-based functional MRI studies. The following emerge as priority areas for improvement.
First, there is a need to incorporate evidence from studies of the trajectories of brain development and studies of brain function. Most addiction neuroscience work has focused on brain function. However, typical measurements of t-fMRI and resting-state functional connectivity are characterized by low heritability, which may arise (at least in part) from reliability challenges. Going forward, outlining the genetic architecture of brain function requires better phenotyping. In addition to improving the reliability of univariate metrics (such as dense-sampling), multivariate approaches may have great utility. Indeed, polyneural phenotypes (that is, those with differentially weighted regional associations, much like polygenic risk scores) may be required to meaningfully predict individual differences in complex behavior. While such ‘lumping’ approaches may disappoint those hoping to link complex behavior and genetic variation to readily interpretable brain regions according to their known role in behavior, they may be replicable and reliable, similar to polygenic approaches. Characterizing the longitudinal trajectories of brain development that contribute to periods of addiction vulnerability is equally important. Efforts targeting brain development, especially large longitudinal samples such as ABCD of middle childhood-young adulthood and the emerging HEALthy Brain and Cognitive Development (HBCD) study, which begins during the prenatal period, will be critical for this process.
Second, as genomic and imaging data become available in larger, population-representative settings that span developmental periods, the GINA model will require retooling to account for lighter and moderate levels of substance use and milder forms of addiction. Distinctions between levels of addiction severity may impact the magnitude of neurogenetic associations or may map onto qualitatively different polygenic signals and brain regions. From a genomics perspective, larger scale discovery GWAS will profoundly impact the GINA model. Currently, far too few GWAS are well-powered to identify genetic signals in individuals of ancestries other than Europeans, which limits the equitable application of PRS. In addition, while smaller studies of copy number variants exist, large-scale meta-analyses of structural variants are needed. Transcriptomic analyses provided some of earliest validation of GWAS discoveries; however, most have been conducted in bulk tissue and greater specificity is needed. Results for other psychiatric disorders have highlighted the importance of studying the enrichment of genetic signals in single cells or single nuclei, thus partitioning heritability (even within a single brain region) into specific cell types. Beyond eQTL, other cell-type specific annotations, such as enhancer effects could also be incorporated into PRS development. Such data are essential to providing a neuro-cellular perspective to the GINA model; however, at this time, larger GWASs are likely to be the most influential source of data. Nonetheless, multi-omics data also provide valuable data matrices for emerging methods for developing functional PRS which appear to be more portable across ancestral populations than traditional effect size based PRS.
A Consideration
Brain neuroimaging and genetics have substantially advanced our understanding of the biological bases of addiction. Both approaches have opponents who have argued that the conceptualization of addiction as a ‘brain disease’ discounts the role of socio-political factors, which may be more modifiable than one’s biology. Yet, influential environmental provocations may in fact not be predictable or malleable. There are also deeper ethical issues surrounding the use of either genetics or neurobiology – or for that matter, environmental factors – to prospectively categorize an individual by their future addiction diagnosis. It is therefore worth considering the tradeoff between using genetics and neuroscience to prognosticate addiction risk as a source of stigma versus the potential for predispositional mechanisms to unburden individuals and societies of ill-construed notions regarding the moral valence of persons using substances.
Summary
Weaving genetics and neuroscience together, especially in the context of environmental considerations, can provide appreciable insights into the origins of addiction. It may even provide clues for preventing the critical transition from non-problematic to maladaptive use and illuminate efficacious therapeutic pathways. By proposing the integrative GINA model, we encourage the adoption of a multifactorial perspective of addiction – a process that represents a dynamic cascade of genetic predisposition and environmental risk and resilience that is enacted via developmentally relevant brain maturation and substance-induced brain alterations.