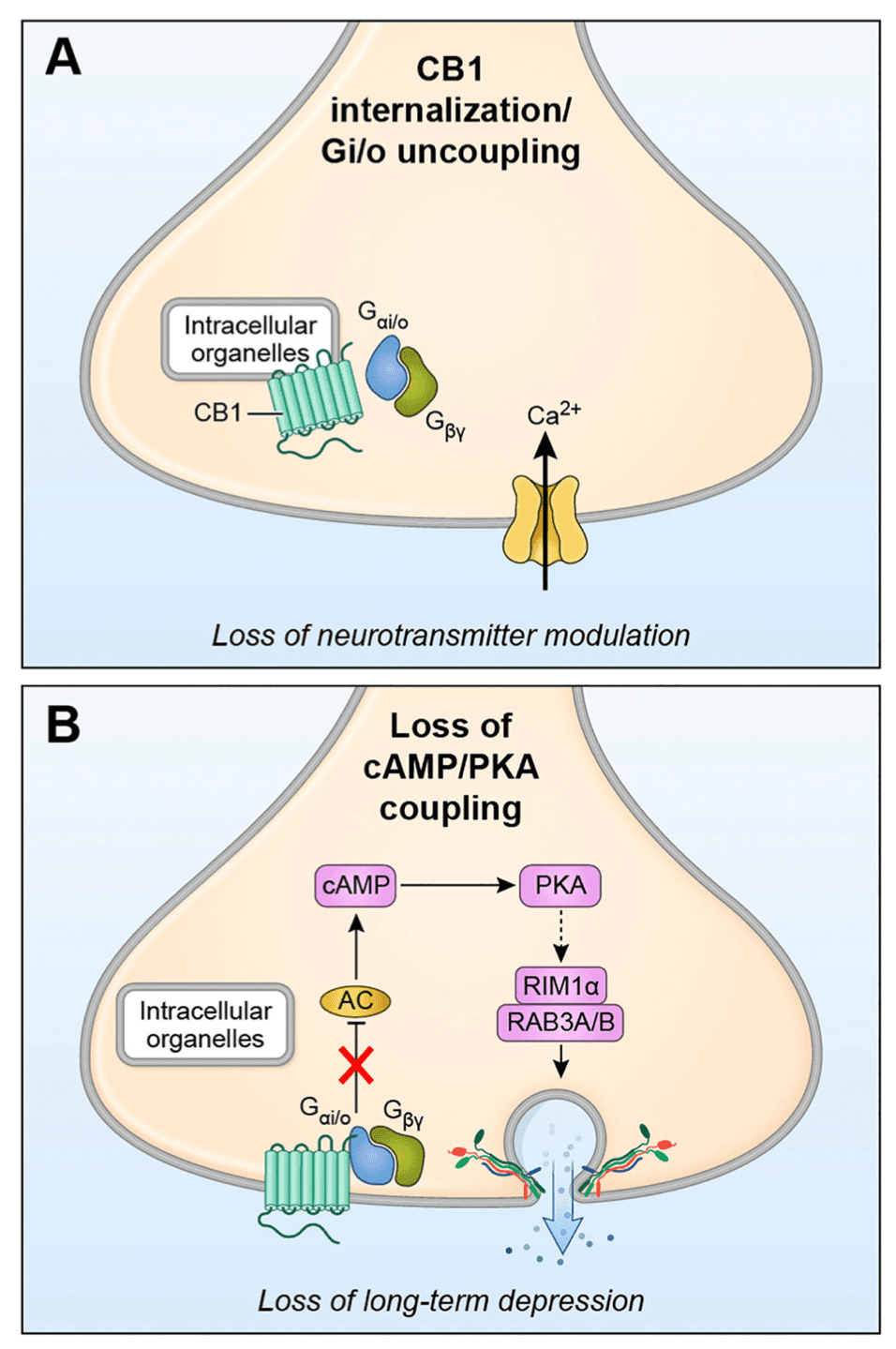
Abstract
The legalization of cannabis in many countries, as well as the decrease in perceived risks of cannabis, have contributed to the increase in cannabis use medicinally and recreationally. Like many drugs of abuse, cannabis and cannabis-derived drugs are prone to misuse, and long-term usage can lead to drug tolerance and the development of Cannabis Use Disorder (CUD). These drugs signal through cannabinoid receptors, which are expressed in brain regions involved in the neural processing of reward, habit formation, and cognition. Despite the widespread use of cannabis and cannabinoids as therapeutic agents, little is known about the neurobiological mechanisms associated with CUD and cannabinoid drug use. In this article, we discuss the advances in research spanning animal models to humans on cannabis and synthetic cannabinoid actions on synaptic transmission, highlighting the neurobiological mechanisms following acute and chronic drug exposure. This article also highlights the need for more research elucidating the neurobiological mechanisms associated with CUD and cannabinoid drug use.
1. Introduction
Cannabis-derived drugs are among the most widely used illicit substances in the world. According to the 2021 World Drug Report, it is estimated that over 200 million people have used cannabis globally, likely increasing amid the global COVID-19 pandemic (World Drug Report 2021, 2021). However, efforts to decriminalize, legalize, and reclassify the scheduling of these drugs are fast transforming the landscape of cannabis use and research. Despite the UN’s reclassification of cannabis and its derivatives, these drugs remain classified as schedule I by the US Drug Enforcement Administration together with other drugs such as heroin, and ecstasy (https://www.dea.gov/drug-information/drug-scheduling). Moreover, the perceived decrease in the risks associated with cannabis use has contributed to its popularity and increased usage, already exacerbating a global health problem (Merrill, 2015).
In addition to cannabis, cannabis derivatives are being developed for medicinal purposes, despite the relative paucity of evidence about their safety, efficacy, and benefits. At the same time the evolving recognition of cannabis use disorder (CUD) and cannabis withdrawal syndrome (CWS) DSM-5; (Association, 2013) complicates the situation (Katz et al., 2014). CUD includes not only tolerance to the drug, but dependence on it as well (Branch et al., 1980; Budney et al., 2007; Hollister, 1978; Hollister, 1986; Jones et al., 1976; Patel and Marwaha, 2019; Zehra et al., 2018) and fits within neurobiological models of substance use disorders (Koob and Volkow, 2016). The intoxicating effects of cannabis consist of euphoria, impaired motor coordination, slowed reaction time, anxiety, impaired judgment/memory, and social withdrawal (Crean et al., 2011; Green et al., 2003; Kowal et al., 2015; Pujol et al., 2014; Solowij and Battisti, 2008; Volkow et al., 2016). Some of these effects can last for hours, depending on the method of cannabis administration (Borodovsky et al., 2016; Carlini et al., 2017; Loflin and Earleywine, 2014; Vandrey et al., 2017). In addition, cannabinoids can increase appetite initially, but chronic use may be suppressive (Cluny et al., 2015; Farokhnia et al., 2020; Foltin et al., 1986; Horn et al., 2018; Kirkham, 2009; Le Strat and Le Foll, 2011; Sansone and Sansone, 2014). Notably, cannabis users are less likely to overdose on cannabis when taken alone compared to other substances of abuse (Crocker et al., 2021; Martins et al., 2015). However, the risk of overdose increases with polysubstance use, such as comorbid use of cannabis with alcohol, opioids, or benzodiazepines (Jordan et al., 2018). In some regular cannabis users, CWS occurs after the cessation of cannabis use and it is a clinical indicator of CUD (Association, 2013). The diagnostic criteria for CWS includes, but is not limited to cannabis craving, sleep disruptions, irritability, aggression, suppressed appetite resulting in weight loss, depression, and anxiety (Karila et al., 2014; Katz et al., 2014; Kesner and Lovinger, 2020; Smith, 2002). CWS can also manifest with physiological symptoms, such as sweating, fever, headaches, fatigue, night sweats, and tremors (Katz et al., 2014). These symptoms can appear within days of cannabis cessation (Davis et al., 2016). There are no approved medications for CUD and CWS. However, behavioral treatments including cognitive and motivational enhancement therapies, as well as contingency management are used by CUD individuals to refrain from cannabis use and to remain abstinent (Sherman and McRae-Clark, 2016). While the behavioral effects of cannabis and withdrawal are well characterized, neurobiological research is just beginning to uncover the mechanisms that contribute to these behavioral changes.
2. Cannabinoids and sites of actions
2.1. Exogenous cannabinoids
The molecular constituents of cannabis-derived drugs (also known as phytocannabinoids) act on several molecular targets within the nervous system and other organs, but the most prominent targets are the Gαi/o protein-coupled cannabinoid receptor types 1 and 2 (CB1 and CB2) (Elsohly and Slade, 2005; Karniol et al., 1975). These receptors inhibit adenylyl cyclase (AC) activity, resulting in decreased levels of cAMP and inhibition of other effector proteins, such as protein kinase activity (PKA) (Ibsen et al., 2017). The predominant psychoactive constituent of cannabis-derived drugs is Δ9-tetrahydrocannabinol (THC), followed by the second most prominent constituent, cannabidiol (CBD). THC acts as a partial agonist at both CB1 and CB2 receptors (Kano et al., 2009; Pertwee, 2008; Pertwee et al., 2010) and its chemical structure was originally identified by Adams and Gaoni in the 20th century (Adams, 1940; Gaoni and Mechoulam, 1971). Within the brain, THC actions on presynaptic CB1 receptors predominately result in inhibitory actions on synaptic transmission (Felder et al., 1995). CB1 receptors are expressed throughout the brain and signaling through these receptors contributes to the behavioral consequences and possibly the misuse liability of cannabinoids (Augustin and Lovinger, 2018; Hu and Mackie, 2015). CB1 receptor signaling has been implicated in the processing and seeking of drug reward (Parsons and Hurd, 2015; Volkow et al., 2017). Unlike THC, which has a high binding affinity to CB1 receptors, CBD has a low binding affinity and may function as a negative CB1/CB2 receptor allosteric modulator (Kathmann et al., 2006; Laprairie et al., 2015; Ryberg et al., 2007). Another molecular constituent of cannabis is cannabinol (CBN), which is produced from the oxidation and degradation of THC (Merzouki and Mesa, 2002; Upton et al., 2013). CBN is a weak CB1 receptor agonist and mildly psychoactive (Rhee et al., 1997). Like THC, CBN binds to CB1 receptors to mediate its drug effect, although at a significantly lower potency than THC (Rhee et al., 1997). This cannabinoid is known to produce a sedative-like feeling (Musty et al., 1976), which may be beneficial to sleep. There is considerable interest in the development of other cannabis-derived drugs, besides THC, as potential therapeutic agents. However, more research is needed to determine their benefits and/or health risk associated with usage.
Full synthetic CB1 receptor agonists are widely used for therapeutic purposes and more recently for recreational drug use as an alternative to cannabis (Waugh et al., 2016; Weinstein et al., 2017). As the name implies, “synthetic” cannabinoids are produced in a laboratory and are chemically distinct from THC. As a result, these compounds are difficult to detect with standard drug screening procedures. Some examples of full CB1 receptor agonists are JWH-018, WIN55, 212–2, and CP55,940 (Atwood et al., 2010; Soethoudt et al., 2017). For a more complete list of synthetic cannabinoids and receptor subtype efficiency see (An et al., 2020). These drugs are pharmacologically similar to THC and exhibit a high binding affinity to CB1/CB2 receptors (Potts et al., 2020; Tai and Fantegrossi, 2017; Walsh and Andersen, 2020). Synthetic cannabinoids are generally more potent than phytocannabinoids, including THC (Banister and Connor, 2018), and are now part of widely abused herbal mixtures referred to as “Black Mamba”, “Spice” or “K2”. Furthermore, these compounds are advertised and/or labeled as “legal” or “fake” cannabis as part of “herbal incense” and “potpourri” to be smoked similarly to cannabis (Kelly and Nappe, 2021; Parrott et al., 2017). These compounds are classified as new psychoactive substances (NPS) (Banister and Connor, 2018; Scourfield et al., 2019) and are often sold online or at gas stations and convenience stores. Unlike THC, these cannabinoids are often not regulated by governments and are easily accessible, especially by adolescents (Johnston et al., 2012). Use of these synthetic compounds can cause drug dependence, tolerance, and withdrawal symptoms, which are all hallmarks of substance misuse (Koob and Volkow, 2016; Nacca et al., 2013; Zimmermann et al., 2009). The neurobiological mechanisms by which these synthetic compounds, such as those in “Spice”, exert their psychoactive effects are not fully understood. Moreover, little is known about the short- and long-term effects of synthetic cannabinoid usage. The use of these compounds raises concerns about escalation of CUD (Alves et al., 2020; Kelly and Nappe, 2021).
2.2. Endogenous cannabinoids
The CB receptors are a key part of the endogenous cannabinoid (endocannabinoid or eCB) juxtacrine signaling system (Ibsen et al., 2017; Lu and Mackie, 2016). The eCBs are naturally produced metabolites of arachidonic acid-containing lipids. These eCBs play important roles in neurodevelopment and synaptic plasticity (Chevaleyre et al., 2006; Lu and Mackie, 2016; Piomelli, 2003). Once released from cells, the eCBs traverse short extracellular distances to activate CB receptors (Mackie, 2005). The two main eCBs are arachidonoyl ethanolamine (AEA) and 2-arachidonoyl glycerol (2-AG). Although both AEA and 2-AG are agonists, 2-AG is a full agonist for CB1/CB2 receptors while AEA is a partial CB1 receptor agonist (Mackie, 2005; Mackie, 2008). These eCBs are synthesized and degraded by distinct enzymatic pathways, which may contribute to differences in physiological and functional roles (Augustin and Lovinger, 2018). In the central nervous system (CNS), eCBs are generated and released mainly from postsynaptic elements such as dendrites, dendritic spines and somata (Alger, 2002; Katona and Freund, 2012; Min et al., 2010). The CB1 receptors are located almost exclusively on presynaptic boutons in CNS where they inhibit neurotransmitter release (Lu and Mackie, 2016).
3. Non-canonical sites of cannabinoid actions
Both exogenous (phyto- and synthetic cannabinoids) and endogenous cannabinoids can bind non-canonical orphan GPCRs, such as G protein-coupled receptor 55 (GPR55) and G protein-coupled receptor 18 (GPR18) (Morales et al., 2017; Morales and Jagerovic, 2016; Pertwee et al., 2010). Additionally, these cannabinoids can bind ligand-gated ion channels and specific transient receptor potential (TRP) channels, as well as nuclear peroxisome proliferator activated receptors (PPARs) (Morales et al., 2017; Muller et al., 2018). For example, the phytocannabinoid CBD interacts with a variety of biomolecules, giving rise to a number of potential physiological actions, albeit although some are indirect (Britch et al., 2021). It is likely that most of the cannabinoid-induced signaling is mediated through the CB1 receptors and these non-canonical GPCRs may play a supporting role in the integration of synaptic signaling to exert their cannabinoid effects. However, more work is needed to determine the functional selectively of these receptors, especially GPR55 and GPR18 (Morales and Jagerovic, 2016).
4. Synaptic effects of cannabinoid drugs
The eCB system is critical in short- and long-term synaptic modulation mainly via the activation of CB1 receptors in the brain. Given the role of eCBs in synaptic transmission, it is likely that eCBs play a role in the optimization and/or refinement of learning, movement control, nociception perception, and stress, either directly or indirectly. Moreover, there is mounting evidence that implicates sex differences in the behavioral and physiological effects of cannabinoids (Borgen et al., 1973; Cocchetto et al., 1981; Craft, 2005; Fattore et al., 2007; Mathew et al., 2003; Wiley, 2003). Exogenous cannabinoids, such as THC and synthetic cannabinoids can disrupt and/or alter eCB signaling and function (Cohen et al., 2020; Deadwyler et al., 1990; Schoeler and Bhattacharyya, 2013). Furthermore, these compounds can trigger different CB1 receptor-mediated intracellular signaling pathways, which may contribute to functional specificity (Laprairie et al., 2014). The effects of acute and chronic cannabinoid drug exposure on synaptic transmission are summarized in Table 1, and discussed further later in the review.
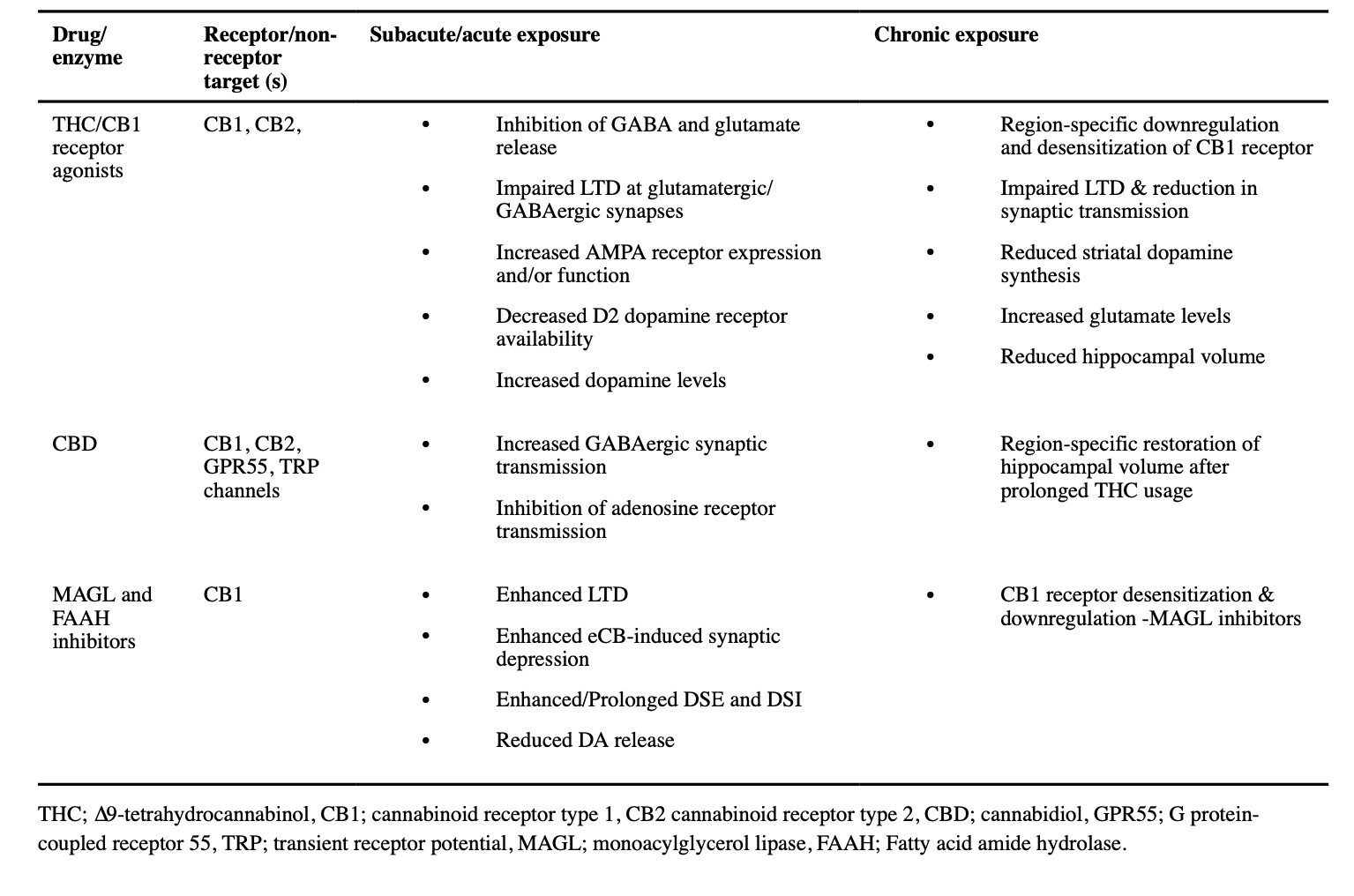
4.1. Acute effects of cannabinoid drugs on synaptic transmission
The actions of full CB1 receptor agonists have been examined at synapses throughout the nervous system. As expected from the localization and coupling of CB1 receptors, activation leads to depression of neurotransmitter release and synaptic responses (Table 1). Presynaptic CB1 receptors are generally coupled to Gi/o-type G-proteins, and receptor activation leads to dissociation of the heterotrimeric G protein subunits (Lu and Mackie, 2016). The Gβ/γ subunits activate the two mechanisms most often implicated in CB1-mediated synaptic depression, inhibition of presynaptic voltage-gated calcium channels (resulting in decreased excitation-secretion coupling) (Fig. 1A), and inhibition of vesicle fusion through actions on the vesicle fusion/release machinery (Fig. 1B) (Chevaleyre et al., 2007; Gerdeman and Lovinger, 2001). The Gα subunit liberated by CB1 activation can inhibit adenylyl AC, leading to decreased signaling by cAMP-activated pathways (Lu and Mackie, 2016). Inhibition of this second messenger system is thought to play a role in long-term synaptic depression (LTD) induced by CB1 activation (Fig. 1C) (Augustin et al., 2014; Chevaleyre et al., 2007; Monday et al., 2018).
These synaptic depressant actions occur mainly at GABAergic and glutamatergic synapses, although release of other neurotransmitters is also affected (Kano et al., 2009). The suppression of inhibitory GABAergic transmission leads to increased synaptic and intrinsic excitability of the recipient postsynaptic neurons. In contrast, suppression of glutamate release will have a net inhibitory effect on postsynaptic neurons. Thus, the effect of a CB1 receptor full agonist in a given brain region depends on the balance of these inhibitory and disinhibitory actions. This balance is, in turn, affected by the locations of the different synapses, the timing of CB1 receptor activation relative to the input from the presynaptic neuron, and receptor expression levels. In the basolateral amygdala, cerebral cortex and hippocampus CB1 receptors are abundant on the axon terminals of one type of GABAergic “basket cell” (BC). These BCs make synapses onto the soma and proximal dendrites of glutamatergic projection neurons. Thus, CB1 receptor activation in these regions will reduce a strong source of inhibition of projection neuron firing, resulting in greater neuronal output. In many brain regions CB1 receptor actions and CB1 expression are especially strong on GABAergic synapses (Adermark and Lovinger, 2009; Katona et al., 2001; Katona et al., 1999; Yoshida et al., 2002).
Activation of CB1 receptors by eCBs has often been shown to enhance induction of long-term potentiation (LTP) at glutamatergic synapses in several brain regions (Carlson et al., 2002). In the basolateral amygdala and hippocampal CA1 regions the disinhibitory CB1 effect induced by inhibition of GABA release is implicated in enhancement of LTP induction at synapses onto projection neurons (Chevaleyre and Castillo, 2004). In the hippocampal CA1 region, depression of GABA release at synapses made by basket cells (Chevaleyre and Castillo, 2003), allows for greater glutamatergic synaptic excitation of pyramidal neurons. This enhances activation of NMDA-type glutamate receptors that are crucial for LTP induction. Similar mechanisms occur in the basolateral amygdala, once again likely involving CB1 receptor activation on GABAergic basket cells (Azad et al., 2004). Similar LTP-enhancing effects of CB1 activation have been observed in other brain regions including striatum (Adermark and Lovinger, 2009).
Levels of eCBs can be enhanced in brain by inhibitors of the enzymes that catalyze their degradation. Fatty acid amide hydrolase (FAAH) is the main enzyme involved in AEA catabolism, and a variety of FAAH inhibitors have been developed (Cravatt et al., 2001; Cravatt et al., 1996; Griebel et al., 2018; Lodola et al., 2015). Application of FAAH inhibitors to brain slices generally does not alter synaptic transmission induced by single afferent stimuli (Table 1). However, in striatum, FAAH inhibition by the URB597 compound inhibits dopamine release induced by repetitive afferent activation (Mateo et al., 2017). This is thought to involve activation of CB1 receptors that reduce glutamate release from cortical terminals. Application of FAAH inhibitors has also been shown to enhance LTD mediated by TRPV1 in nucleus accumbens (NAc) brain slices, presumably by increasing AEA activation of this receptor (Grueter et al., 2010). In the hippocampal dentate gyrus, URB597 application has no effect on glutamatergic synaptic transmission, but inhibition is observed when combined with AEA administration (Chavez et al., 2010). This is presumably because the FAAH inhibitor prevents degradation of the applied AEA. Activation of TRPV1 receptors by AEA appears to be the mechanism underlying this synaptic depression. The catabolic enzyme for 2-AG is monoacylglycerol lipase (MAGL). Inhibition of MAGL by the JZL184 compound inhibits repetitive stimulus-driven dopamine release, as previously described for FAAH inhibition (Mateo et al., 2017). Inhibitors of MAGL have little effect on synaptic transmission when applied alone, but prolong eCB-mediated depolarization-induced suppression of inhibitory and excitatory transmission (DSE and DSI respectively) (Hashimotodani et al., 2007; Pan et al., 2009; Straiker et al., 2009; Szabo et al., 2006). This is one key piece of evidence indicating that 2-AG mediates the synaptic depression observed in this experimental paradigm. Interestingly, similar effects are seen at some synapses with inhibitors of the COX-2 enzyme that can also contribute to 2-AG degradation (Straiker et al., 2011; Yang et al., 2008)
Impairment of LTP has been observed during application of full CB1 agonists to brain slices (Table 1). The initial characterization of this effect was in brain slices from prefrontal cortex, where application of a full CB1 agonist reduced the incidence of LTP at synapses onto projection neurons, while increasing the incidence of LTD (Auclair et al., 2000). Subsequent studies of LTP in the hippocampal CA1 subregion produced similar findings, in this case during application of highly efficacious CB1 agonists found in spice-like drugs (Basavarajappa and Subbanna, 2014; Hoffman et al., 2017). This finding suggests that activation of these CB1 receptors at or near maximal efficacy has effects different from partial receptor activation. One possible mechanism underlying LTP disruption by CB1 activation is inhibition of glutamatergic transmission that is needed for LTP induction (Bajo et al., 2009; Slanina et al., 2005).
The synaptic effects of THC have been studied (Table 1), but not to the same extent as full CB1 agonists, mainly due to difficulty in obtaining this highly controlled substance and the very low solubility of this compound (Hoffman and Lupica, 2013). Early studies examined effects of applying THC to brain slices. A biphasic concentration-response was observed in the hippocampal CA1 region, with potentiation of a field potential population spike (PS) and prolongation of the duration of LTP at low-moderate concentrations, changing to inhibition of the PS and shortening of LTP duration at higher concentrations (Foy et al., 1982; Nowicky et al., 1987). However, it was difficult to ascertain the synaptic changes underlying these THC actions, as only extracellular field potential recordings were performed. When GABAergic IPSCs are measured with whole-cell recording in hippocampal CA1 slices, inhibition by THC is comparable to that of full CB1 agonists (Laaris et al., 2010). These findings are consistent with findings indicating that CB1 receptors are most abundant on GABAergic terminals in hippocampal CA1 (Katona et al., 1999). However, it is notable that THC also produces substantial inhibition of glutamatergic synaptic responses at Schaffer collateral-CA1 pyramidal neurons in hippocampal slices (Hoffman et al., 2010). Strong inhibition of glutamatergic transmission by THC has also been observed in hippocampal cultures (Shen and Thayer, 1999), and striatal slices (Brown et al., 2003). However, it should be noted that Straiker and Mackie (2005) did not observe inhibition of glutamatergic transmission in hippocampal autaptic cultures but did find that THC blocked the effect of other CB1 agonists. Thus, despite the fact that THC is a partial CB1 agonist it can have profound effects on both excitatory and inhibitory synaptic transmission. Other synaptic sites of THC action have been suggested, including ionotropic glycine receptors (Hejazi et al., 2006; Xiong et al., 2011).
The acute synaptic effects of CBD have been the subject of increasing research activity in recent years (Table 1). One proposed molecular CBD target is the G protein-coupled lysophosphatidylinositol receptor known as GPR55 (Ryberg et al., 2007). In hippocampal brain slices CBD enhances GABAergic synaptic responses in dentate gyrus (DG) granule neurons (Kaplan et al., 2017). This effect is mimicked and prevented by the GPR55 antagonist DIC 16020046, supporting a role for antagonism of this receptor in the CBD action. Other synaptic receptors implicated in CBD action include the 5-HT-1A receptor (Rock et al., 2012; Russo et al., 2005), the ionotropic glycine receptor (Ahrens et al., 2009), and several transient receptor potential (TRP) ionotropic channels (De Petrocellis et al., 2011). Depending on the TRP channel subtypes, CBD can have varying effects on channel activation. For example, TRPV1 and 2, as well as TRPA1 are activated by CBD, whereas CBD acts as an antagonist at TRPM8 (Bisogno et al., 2001; De Petrocellis and Di Marzo, 2010; De Petrocellis et al., 2008; Muller et al., 2018; Qin et al., 2008). The CBD actions involving the 5-HT1A receptor have been suggested to underlie in vivo actions of the drug, including potential uses in treatment of Parkinson’s Disease symptoms (Patricio et al., 2020). There is also evidence that CBD can inhibit adenosine uptake (Pandolfo et al., 2011). However, there is little data indicating if CBD alters synaptic transmission mediated by these receptors in in situ preparations or the intact brain. The synaptic effects of CBD may contribute to the antiepileptiform and antiseizure effects of the drug (Jones et al., 2010; Rosenberg et al., 2015). The related cannabis constituent cannabidivarin has also been shown to activate TRP channels (Iannotti et al., 2014). This compound also inhibits epileptiform activity in hippocampal slices, and this effect in intact tissue appears to involve TRPV1.
4.2. Subacute CB1 agonist actions
The “subacute” effects of THC and other cannabinoid drugs are defined herein as synaptic changes observed within hours of a few days following a single in vivo drug exposure. The most commonly used paradigm is a single intraperitoneal drug injection followed some hours later by brain slice preparation and electrophysiological recording (Table 1). For example, Mato and coworkers observed that LTD was impaired at GABAergic synapses in the CA1 hippocampal subregion 15–20 hours after a single THC injection (Mato et al., 2004). These investigators also found a similar impairment of LTD at glutamatergic synapses in the NAc. Impaired LTD persists for up to three days after the drug injection. In slices from the ventral tegmental area (VTA), an increase in synaptic AMPA receptors was observed 24 hours following a single THC injection, perhaps indicative of long-term potentiation at these synapses (Good and Lupica, 2010).
5. Chronic effects of cannabinoid drugs on synaptic transmission
Chronic cannabinoid drug use is associated with increased risk of developing CUD, which is accompanied by neurobiological changes in reward-related and cognitive circuit functions. Cognitive changes following chronic cannabinoid use contribute in part to the maintenance of CUD via increased impulsivity and impaired decision making (Crean et al., 2011; Irimia et al., 2015). Tolerance to the effects of cannabinoid drugs is another notable phenotype that has been widely examined in animal models (Maldonado and Rodriguez de Fonseca, 2002; Martin et al., 2004; Sim-Selley, 2003). The most consistent neurobiological consequence of chronic THC exposure is decreased CB1 receptor numbers, as well as decreased CB1 agonist and eCB-induced presynaptic modulation (reviewed in (Hoffman et al., 2021; Kesner and Lovinger, 2020)) (Table 1). Long-term exposure to THC and synthetic cannabinoids results in region-specific decreases in CB1 receptor expression which likely contribute to differences in receptor radioligand binding and reduced drug sensitivity (Breivogel et al., 1999; Corchero et al., 1999; Oviedo et al., 1993; Rodriguez de Fonseca et al., 1994; Romero et al., 1998; Romero et al., 1997; Zhuang et al., 1998). Furthermore, chronic exposure to cannabinoids impairs synaptic plasticity, and this impairment is accompanied by the desensitization of CB1 receptors (Hoffman et al., 2003; Mato et al., 2005). The desensitization and downregulation of CB1 receptors have been consistently implicated in the development of cannabis tolerance (Maldonado, 2002; Martin et al., 2004; Sim-Selley, 2003) and have been observed in individuals that regularly use cannabis (Villares, 2007). Chronic cannabinoid exposure produces widespread reversible downregulation and desensitization of CB1 receptors (Sim-Selley, 2003). The magnitude of downregulation is generally 25–50% depending on the measurement type (radioligand binding, immunochemistry or signaling) and brain region examined. Greater decreases were reported in the cerebellum and hippocampus compared to somewhat smaller magnitudes in the basal ganglia and midbrain (Breivogel et al., 1999; Cichewicz et al., 2001; Sim-Selley, 2003; Sim-Selley et al., 2006). Given that the magnitude of down-regulation is smaller in the midbrain and basal ganglia, it is not surprising that the rate of reversal is faster in the striatum and midbrain than in other brain regions (Sim-Selley et al., 2006). Together, these mechanisms provide the underlying biological properties by which cannabinoid tolerance can develop (Maldonado et al., 2011; Maldonado and Rodriguez de Fonseca, 2002; Tanda and Goldberg, 2003).
The functional significance of AEA and 2-AG in promoting cannabinoid tolerance and dependence have been explored. Repeated administration of AEA or an AEA analog (methanandamide) has yielded inconclusive findings on the reduction of CB1 receptor density and signaling (Aceto et al., 1998; Romero et al., 1999; Romero et al., 1995; Rubino et al., 2000a). Furthermore, pharmacological blockade of FAAH does not alter CB1 receptor expression and function (Schlosburg et al., 2010). In mice lacking FAAH, chronic administration of THC produces similar magnitudes of CB1 receptor desensitization and downregulation in the striatum, hippocampus, and cerebellum compared to controls (Falenski et al., 2010). These findings suggest that AEA signaling doesn’t contribute to the receptor downregulation in response to chronic THC exposure. It is worth mentioning that the deletion of FAAH does not alter CB1 receptor expression in brain (Cravatt et al., 2001). On the other hand, chronically elevated levels of 2-AG achieved by genetic deletion or pharmacological blockade of MAGL results in the reduction of CB1 receptor density and function (Chanda et al., 2010; Schlosburg et al., 2010). Thus, increased 2-AG tone can possibly desensitize and down-regulate CB1 receptors (Table 1).
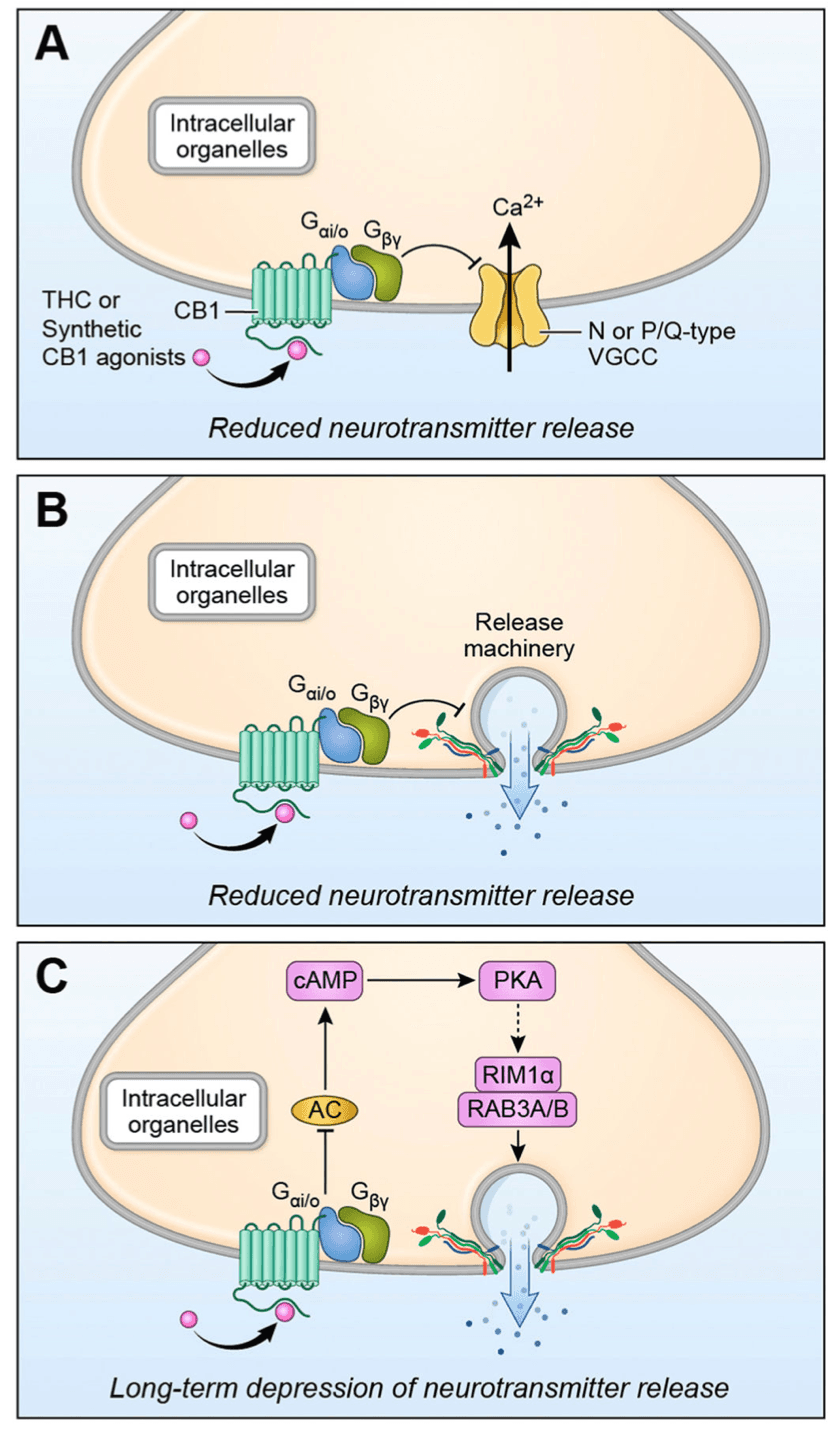
The molecular mechanisms involved in CB1 receptor desensitization and downregulation have been explored, but to date there is little evidence about which mechanisms are involved in loss of presynaptic receptor function. The radioligand binding studies mentioned above, and a few immunochemical studies that indicate decreased receptor numbers are consistent with CB1 receptor internalization or degradation as candidate mechanisms for CB1 receptor desensitization (reviewed in (Hoffman et al., 2021; Kesner and Lovinger, 2020)) (Fig. 2A). Indeed, loss of cell surface receptors consistent with these mechanisms has been observed in heterologous cellular expression systems (Hsieh et al., 1999; Rinaldi-Carmona et al., 1998; Tappe-Theodor et al., 2007) and in primary neuronal cultures (Coutts et al., 2001; Tappe-Theodor et al., 2007). The most direct evidence of decreased CB1 receptor on the plasma membrane of presynaptic terminals induced by chronic THC exposure in adult animals was obtained with super-resolution microscopy imaging or GABAergic terminals in the hippocampal CA1 region (Dudok et al., 2015). Recovery of terminal surface CB1 levels was observed several weeks after cessation of THC treatment. However, reduced CB1 levels are not always apparent under conditions where decreased receptor induced signaling and tolerance to agonist effects on transmission are observed (Romero et al., 1999; Romero et al., 1998; Romero et al., 1997; Rubino et al., 2000b). Thus, other mechanisms of desensitization, such as uncoupling of CB1 receptors from G proteins and other intracellular signaling mechanisms, or changes in the intracellular signaling systems themselves have also been explored (Fig. 2B) (Hutcheson et al., 1998; Martin et al., 2004; Rubino et al., 2000b; Sim-Selley, 2003). As yet, there is insufficient evidence as to which of these mechanisms underlie the loss of eCB/CB1-mediated plasticity following chronic THC or agonist exposure.
Chronic exposure to THC and CB1 receptor agonists impairs synaptic LTD at glutamatergic synapses in the dorsal and ventral striatum, areas of the brain important for eCB-mediated habitual actions and reward processing, respectively (Augustin and Lovinger, 2018; Chen et al., 1990; Herkenham et al., 1991; Hoffman et al., 2003; Hoffman et al., 2007; Nazzaro et al., 2012; Robbe et al., 2001) (Table 1). The NAc/ventral striatum integrates incoming sensory inputs to modulate cortical activity to influence drug seeking behavior. In fact, drug-induced impairments in LTD have been implicated in compulsive drug seeking and/or taking behavior (Kasanetz et al., 2010). Not surprisingly, operant self-administration of THC and CBD inhibits the induction of LTD at cortical inputs to NAc (Neuhofer et al., 2020), again, bolstering the argument that cannabinoid reward-driven signaling, resulting in voluntary drug seeking/taking behavior is mediated by the inhibition of glutamatergic signaling at these synapses.
Little is known about the effects of CBD alone on long-term changes in synaptic plasticity. However, CBD is hypothesized to reduce the aversive effects of THC (Greydanus et al., 2013; Klein et al., 2011; Russo and Guy, 2006; Vann et al., 2008). It is likely that CBD may exert its long-term effects on plasticity at non-CB1/CB2 receptors, given its low affinity for these receptors (Zou and Kumar, 2018).
The loss of NAc LTD is accompanied by the desensitization of CB1 receptors following chronic THC and cannabinoid exposure (Mato et al., 2005; Neuhofer et al., 2020). Differential changes in the signaling strength of these inputs may contribute to the reinforcing properties of cannabinoid drugs and the regulation of behavior. Alterations in dendritic spine density and morphology in NAc medium spiny neurons occur following chronic exposure to THC (Kolb et al., 2006; Kolb et al., 2018; Spencer et al., 2018). Moreover, loss of LTD is observed at synapses originating from the medial prefrontal cortex (mPFC) and ventral hippocampus (vHipp) (Hwang and Lupica, 2020). Interestingly, chronic THC exposure during adolescence inhibits LTD induction in the mPFC, which is reversed by enhancing AEA levels (Bara et al., 2021; Cuccurazzu et al., 2018). Collectively, these results support the idea that cannabinoids alter CB1 receptor-mediated synaptic modulation to facilitate the reinforcing effects of these drugs and promote tolerance and dependence. Interestingly, chronic THC exposure results in synapse-specific LTD impairments in the NAc, in which LTD at mPFC and vHipp synapses, and not LTD at synapses originating from the basolateral amygdala (BLA) are affected (Hwang and Lupica, 2020). These results suggest that THC-induced synaptic modifications may have very different effects on striatal output, depending on the MSN synapses affected. However, more research is needed to identify whether the induction of LTD at synapses onto different MSN subtypes, indirect- and direct-projecting, are affected. In addition to these presynaptic changes, there is a postsynaptic strengthening of the BLA and vHipp inputs, and the weakening of inputs from mPFC (Hwang and Lupica, 2020). These changes in presynaptic and postsynaptic mechanisms of glutamatergic signaling highlight the importance of the synaptic balance among these brain regions to guide behavior. After prolonged exposure to THC, there is a shift from cortical regions involved in cognitive processes to subcortical regions, which are involved in memory and emotional processes to promote drug seeking and dependence (Hwang and Lupica, 2020; Lafferty and Britt, 2020). Additionally, it may be this dysregulation in synaptic transmission that contributes to the cannabinoid-induced deficits associated with memory formation.
Loss of LTD in dorsolateral striatum (DLS) following chronic THC exposure appears to contribute to development of habitual instrumental behavior (Nazzaro et al., 2012). The loss of this presynaptic modulation may lead to increased efficacy of inputs from sensorimotor cortical regions that drives DLS and the associated basal ganglia circuitry supporting stimulus/context-driven behavior that overrides goal-direct actions. Synaptic depression mediated by eCBs and CB1 also plays a role in habitual behavior through suppression of orbitofrontal cortical inputs to dorsomedial striatum (Gremel et al., 2016). In this case, the eCB-mediated synaptic modulation suppresses cortical input that drives goal-directed behavior, favoring habitual responding. It has yet to be determined how chronic exposure to THC or other CB1 agonists alters this dorsal striatal eCB role.
Functional cannabinoid tolerance also occurs at excitatory and inhibitory synapses in the hippocampus (Bolla et al., 2002; Hampson and Deadwyler, 1999; Pope Jr et al., 2002). CB1 receptors modulate both glutamatergic and GABAergic hippocampal transmission (Carlson et al., 2002; Chevaleyre and Castillo, 2004). However, these receptors are more abundantly expressed on GABAergic rather than glutamatergic terminals (Katona and Freund, 2008; Marsicano and Lutz, 1999). Modifications at these synapses are likely to contribute to the long-lasting cannabinoid-induced deficits in memory and cognition. Chronic exposure to THC blocks LTP at glutamatergic synapses in the dentate gyrus and CA1 regions of the hippocampus via a CB1 receptor-mediated mechanism (Fan et al., 2010; Hoffman et al., 2007). Similar impairments in hippocampal CA1 LTP are observed in vivo with repeated exposure to HU-210, a CB1 receptor full agonist (Hill et al., 2004). Moreover, these impairments are associated with deficits in working memory. It is likely that reduction in glutamate receptor expression and function contributes to the impairments in hippocampal synaptic plasticity (Fan et al., 2010). Chronic exposure to THC reduces cannabinoid CB1 receptor sensitivity and/or efficacy at GABAergic, but not glutamatergic terminals in the CA1 region (Hoffman et al., 2007). This reduced sensitivity is accompanied by a downregulation of CB1 receptors, likely contributing to reduced receptor sensitivity (Dudok et al., 2015). In the hippocampus, eCBs facilitate the induction of LTP through the transient suppression of GABAergic transmission, as mentioned previously (Carlson et al., 2002; Chevaleyre and Castillo, 2004; Monory et al., 2015). However, after chronic THC exposure, the suppression of GABAergic inhibition onto glutamatergic synapses is lost, possibly resulting in LTP impairments. However, this has not been directly tested. Repeated THC exposure also results in the reduction of dendritic spine density in CA1 hippocampal neurons (Chen et al., 2013). These results demonstrate that chronic cannabinoid exposure can induce cellular and synaptic changes that alter plasticity to affect behaviors, such as spatial learning and working memory.
Generally, drugs of abuse often impair the induction of LTP and LTD in reward-related drug seeking and taking circuits (Abrahao et al., 2017; Belmeguenai et al., 2008; Kauer and Malenka, 2007; Martin et al., 2006; Shen and Kalivas, 2013; Wolf et al., 2004). Within the ventral tegmental area (VTA), chronic THC or CB1 agonist induces LTD at glutamatergic terminals that is dependent on NMDA and AMPA GluA2 receptor endocytosis and CB1 receptor signaling (Liu et al., 2010). The VTA is an integral brain region involved in the reward pathway and sends projections to NAc and mPFC, brain regions involved in regulating the cognitive and motivational aspects of behaviors (Sesack and Grace, 2010).
Chronic drug use can result in long-term changes in synaptic plasticity that persist long after drug cessation. LTD impairments are observed following the self-administration of THC and CBD and a withdrawal period (Neuhofer et al., 2020; Spencer et al., 2018), as well as after chronic exposure (7 days) of THC (Hoffman et al., 2007). It is reported that CB1 receptor downregulation can persist for a short period of time following termination of cannabinoid exposure (Sim-Selley et al., 2006). Some of the neurobiological alterations that occur with prolonged cannabinoid use are reversible. More work is needed to determine which of these changes are reversible or how long this reversal process might take. For example, in human cannabis users, CB1 receptor binding function returns to normal levels after a period of abstinence (D’Souza et al., 2016; Hirvonen et al., 2012). There is also a partial recovery of LTP after the cessation of THC exposure in rodents, suggesting that the cognitive and reward-processing deficits are not permanent (Hoffman et al., 2007). The overall net effects of chronic cannabinoid drug use are the loss of synaptic plasticity that may contribute to tolerance and stabilize drug-oriented behaviors.
6. Cannabis use effects on synaptic plasticity in human brain
Only one study to date has examined synaptic plasticity in the brain of humans diagnosed with CUD (Martin-Rodriguez et al., 2021). In this study, continuous theta burst stimulation (cTBS) delivered via transcranial magnetic stimulation of the motor cortex was used to examine plasticity of motor evoked potentials (MEPs) measured in a contralateral muscle. Decreased MEP persisting for 10s of minutes was induced by cTBS in non-cannabis-users and in cannabis users that did not meet CUD criteria. In contrast this decrease was not observed in individuals with CUD. This finding indicates that high levels of cannabis use can reduce plasticity in the human brain. A reduction in hippocampal volume has been reported in long-term heavy cannabis users compared to non-cannabis users (Demirakca et al., 2011; Yucel et al., 2016; Yucel et al., 2008) (Table 1). This reduction is likely to contribute to alternations in functional connectivity and cognitive impairments associated with chronic cannabis use (Broyd et al., 2016; Harding et al., 2012). Interestingly, prolonged CBD usage has been implicated in restoring hippocampal volume in a region-specific manner and improving cognitive function in chronic cannabis users (Beale et al., 2018; Solowij et al., 2018) (Table 1). More studies examining THC, CB1 agonist effects, and other cannabis derivatives on plasticity, as well as alterations in plasticity and acute drug effects on plasticity in individuals with CUD would help in assessing the translatability of findings in laboratory animals and in back-translating studies from humans to animal models.
7. Cannabis use effects on neurotransmitter levels in human brain
There is limited evidence to date of the effects of acute and chronic cannabis use on markers of synaptic function in human brain, but imaging techniques have provided some intriguing findings. Ligands for positron emission tomography (PET)-based imaging of CB1 receptors have been developed over the last 10–15 years (Hirvonen, 2015). Studies employing these PET ligands in humans have revealed evidence of a small but significant decrease in CB1 receptor availability in several cortical and limbic brain regions (Ceccarini et al., 2015; Hirvonen et al., 2012; Spindle et al., 2021; Weinstein et al., 2016). As mentioned above, the receptor levels return to near baseline values following cessation of cannabis use (Hirvonen et al., 2012).
Many psychoactive drugs, including all drugs that give rise to SUDs, alter brain dopamine (DA) levels (Volkow and Morales, 2015). In general, acute exposure to these drugs increases DA release in the NAc and other brain regions implicated in reward and behavioral actions of these drug (Volkow and Morales, 2015). Cannabis and THC have similar acute DA-increasing actions thought to arise from decreased inhibition of midbrain dopaminergic neurons (Lupica and Riegel, 2005; Peters et al., 2021). Thus, it is important to understand cannabis effects on DA in the human brain. To this end, several recent studies have used brain imaging and spectroscopy approaches to estimate changes in brain DA levels, DA receptor density and other molecules involved in dopaminergic transmission following acute of chronic cannabis use.
A few studies have examined availability of D2 receptors in human striatum using PET or single-photon emission tomography (SPET) imaging to determine if acute THC administration alters binding to striatal D2 dopamine receptors, which is one way to estimate DA release in human brain (Barkus et al., 2011; Bossong et al., 2015; Bossong et al., 2009; Stokes et al., 2009). Bossong and coworkers observed a modest decrease in receptor availability in ventral striatum, consistent with increased DA release, that correlated with THC plasma concentration (Bossong et al., 2015). The other studies found no evidence for altered release despite clear psychoactive effects of the administered THC. These inconclusive results may be due to the relatively low sensitivity of this indirect approach and smaller sample sizes in the (Barkus et al., 2011) and (Stokes et al., 2009) studies.
Using PET imaging to measure DA synthesis capacity with the precursor 18F-DOPA, Bloomfield and coworkers found evidence of reduced DA synthesis in striatum of regular cannabis users (Bloomfield et al., 2014) (Table 1). The whole striatal dopamine synthesis capacity was correlated with the rate of cannabis use as well as the age of onset of first cannabis exposure in this study, with early onset and heavy drug associated with greater synthesis impairment. No correlation between DA synthesis rate and psychotic symptoms was observed. Similar conclusions were drawn in studies examining decreased D2-like dopamine receptor availability following amphetamine and methylphenidate administration in cannabis users determined to be dependent on the drug (van de Giessen et al., 2017; Volkow et al., 2014). Binding availability of the dopamine transporter (DAT) was also reduced in individuals who regularly use both tobacco and cannabis (Leroy et al., 2012). However, it is not clear that use of cannabis, as opposed to tobacco, contributed to this effect. Changes in DA release in response to a stressful task were not altered in regular cannabis users (Mizrahi et al., 2013). Decreased availability of D2-like dopamine receptors has been touted as a biomarker of several substance use disorders (Volkow and Morales, 2015). However, in PET studies of regular cannabis users no change in D2-like availability was detected (Urban et al., 2012), and similar findings were obtained by (van de Giessen et al., 2017) and (Volkow et al., 2014). Thus, there is no evidence that decreased D2-like receptor levels are associated with regular cannabis use or CUD. Overall, studies of cannabis and the dopamine system in humans indicates that acute drug-induced increases in DA are smaller than those produced by other drugs of abuse. Chronic cannabis use, including use that meets the criteria for CUD, appears to result in decreased dopaminergic function, but there is no decrease in D2 receptor levels, unlike those associated with use disorders for other drugs. It is likely that CUD is associated with relatively mild disruption of brain dopaminergic systems and thus it is not yet clear if DA plays a strong role in this use disorder.
Effects of cannabis on levels of glutamate in human brain have been examined using magnetic resonance spectroscopy (MRS). Administration of THC induced an increase in glutamate levels in the striatum, and the caudate nucleus, in particular (Colizzi et al., 2020; Mason et al., 2019). Lower baseline glutamate levels and larger THC-induced increases were observed in participants in which THC had psychotomimetic effects (Colizzi et al., 2020). Increased striatal glutamate levels were also associated with decreased connectivity of limbic striatum and cortical subregions as measured by functional magnetic resonance imaging (Mason et al., 2019). No effect on GABA concentrations was observed in this study. One study that examined glutamate levels in individuals with CUD found no differences in the levels of the neurotransmitter in hippocampus or striatum compared to the control group (van de Giessen et al., 2017). These findings indicate that THC-induced increases in striatal glutamate may contribute to alterations in brain circuitry associated with cognitive dysfunction and psychosis, but clearly more work is needed on the effects of regular, heavy cannabis use.
Both DA and glutamate are implicated in endocannabinoid-mediated LTD in striatum (Lovinger and Mathur, 2012). Dopamine also plays a key role in LTP at glutamatergic synapses in striatum (Calabresi et al., 2007). Thus, it will be of interest to determine how cannabis use and THC exposure alter striatal synaptic plasticity and associated forms of learning and memory (Augustin and Lovinger, 2018).
8. Conclusions
Agonists for the CB1 receptor, including THC, produce acute intoxication. Long-term CB1 agonist administration at high doses can lead to CUD. The CB1 receptors normally participate in eCB-mediated short- and long-term synaptic plasticity, including both LTD and LTP. Acutely, the CB1 agonists override eCB modulation causing disruption of physiological synaptic plasticity processes. It has long been appreciated that THC is a partial agonist at CB1, while other naturally occurring and synthetic compounds exhibit a range of efficacies. The CB1 partial agonists would be expected to produce smaller effects on intracellular signaling and synaptic transmission than full agonists, and this is evident in studies using GTPgammaS binding (Griffin et al., 1999). The efficacy of AEA for inhibiting voltage-gated calcium channels and activating inwardly-rectifying potassium channels is similar to that of 2-AG and full agonists (Guo and Ikeda, 2004; Mackie et al., 1995; Twitchell et al., 1997), although it should be noted that AEA effects may be partially mediated by a non-CB1 mechanism (Guo and Ikeda, 2004). However, effects on synaptic transmission are not as clear. The full synthetic agonists and 2-AG generally produce robust synaptic depression, while noladin ether and AEA have smaller effects (Straiker and Mackie, 2005). As discussed above, the actions of THC are inconsistent, with inhibition occurring at some synapses, but not at others (Brown et al., 2003; Hoffman et al., 2010; Shen and Thayer, 1999; Straiker and Mackie, 2005). This differential efficacy could reflect differences in receptor reserve (e.g. due to differential CB1 density) on different presynaptic terminals, with THC efficacy expected to be larger at synapses with greater reserve. It must also be noted that partial agonists can prevent/occlude effects of full agonists by occupying binding sites.
Chronic exposure to THC and other CB1 agonists leads to loss of presynaptic CB1 actions in several brain regions. This loss of receptor function leads, in turn, to deficient eCB-mediated synaptic plasticity that disrupts cognitive functions and contributes to altered reward processing and CUD. Disruption of plasticity and alteration in neurotransmitter levels following CB1 agonist exposure has now been observed in both animal models and human volunteers. In contrast to THC and synthetic high-efficacy CB1 agonists, cannabis constituents such as CBD and CBN have weak actions at CB1 receptors and are not intoxicating or associated with CUD. It remains to be determined how these compounds alter brain function, although several mechanisms have been proposed for CBD. While candidate molecular mechanisms have been identified for loss of CB1 function following chronic agonist exposure, more work is needed to determine if and where these mechanisms occur at presynaptic terminals throughout the brain. Further research is also needed to determine effects of chronic CB1 agonist exposure on brain circuit function, and the contribution of these actions to CUD.