Abstract
Psychedelics produce fast and persistent antidepressant effects and induce neuroplasticity resembling the effects of clinically approved antidepressants. We recently reported that pharmacologically diverse antidepressants, including fluoxetine and ketamine, act by binding to TrkB, the receptor for BDNF. Here we show that lysergic acid diethylamide (LSD) and psilocin directly bind to TrkB with affinities 1,000-fold higher than those for other antidepressants, and that psychedelics and antidepressants bind to distinct but partially overlapping sites within the transmembrane domain of TrkB dimers. The effects of psychedelics on neurotrophic signaling, plasticity and antidepressant-like behavior in mice depend on TrkB binding and promotion of endogenous BDNF signaling but are independent of serotonin 2A receptor (5-HT2A) activation, whereas LSD-induced head twitching is dependent on 5-HT2A and independent of TrkB binding. Our data confirm TrkB as a common primary target for antidepressants and suggest that high-affinity TrkB positive allosteric modulators lacking 5-HT2A activity may retain the antidepressant potential of psychedelics without hallucinogenic effects.
Incidence of depression has surged during the past decade especially among young individuals1. There is therefore an urgent need for new, more efficient treatments for depression. Preliminary clinical trials suggest that psychedelics lysergic acid diethylamide (LSD) and psilocybin, through its metabolite psilocin (PSI), hold promise as fast-acting antidepressants with long-lasting therapeutic effects that are at least as effective as those of currently used antidepressants. Until now, the acute hallucinogenic effects of psychedelics through activation of the serotonin 2A receptor (5-HT2A) have restricted their widespread clinical use, as they require specialized medical supervision during prolonged sessions set in a controlled clinical environment. In addition, concerns exist that psychedelics may trigger hallucinogen persisting perception disorder (HPPD) or irreversible episodes of psychosis in susceptible populations, which has already led to the exclusion of patients with a family history of bipolar disorder or schizophrenia from participating in psychedelic clinical trials for depression. However, recent reports suggest that the hallucinogenic effects of psychedelic compounds can be separated from their antidepressant-like and plasticity-promoting effects, indicating that it may be possible to find compounds or treatment combinations that retain the antidepressant effects of psychedelics, but are devoid of the hallucinogenic effects. It is, however, unclear how these separate effects are mediated.
Essentially all antidepressant drugs, including psychedelics, promote neuroplasticity, which is considered a critical component of their therapeutic effect. Brain-derived neurotrophic factor (BDNF) and its receptor TrkB (neurotrophic receptor tyrosine kinase, Ntrk2) are central mediators of plasticity and the therapeutic action of antidepressants. Recent findings show that antidepressants, including conventional antidepressants such as fluoxetine and imipramine, as well as the rapid-acting ketamine, directly bind to TrkB and allosterically potentiate BDNF signaling. BDNF and TrkB have also been implicated in the action of psychedelics as downstream effectors of 5-HT2A activation. In this Article, because psychedelics produce robust spinogenesis and dendritogenesis and these effects are known to require intact TrkB signaling, we set out to explore whether direct binding to TrkB might mediate the neuroplastic effects behind their therapeutic potential.
Results
Psychedelics are high-affinity TrkB ligands
We first tested whether psychedelics bind to TrkB in lysates of HEK293T cells transiently expressing TrkB and immunoprecipitated with a TrkB antibody. We observed that radiolabeled LSD (3H-LSD) binds directly to human (Kd = 0.930 ± 0.414 nM, Bmax = 4.028 ± 0.326 pmol mg−1 protein), rat (Kd = 0.656 ± 0.093 nM, Bmax = 3.053 ± 0.099 pmol mg−1 protein) and mouse TrkB (Kd = 0.425 ± 0.296 nM, Bmax = 7.388 ± 0.771 pmol mg−1 protein) (Fig. 1a and Extended Data Fig. 1a,b,d) with high affinity, which is similar to that for its canonical target 5-HT2A (Ki = 3.5 nM). Remarkably, the affinity of LSD to TrkB was up to 1,000-fold higher than that of other antidepressants such as fluoxetine and ketamine to TrkB. To verify the binding and to localize it, we tested the binding of 3H-LSD to a set of TrkB mutants (Fig. 1a). Replacement of the transmembrane domain (TMD) of TrkB with the corresponding sequence of TrkA (TrkA.TM) completely abolishes LSD binding, which restricts the LSD binding site to the TMD of TrkB. A tyrosine-to-phenylalanine point mutation (Y433F) in the TMD of TrkB that disrupts binding and plasticity-related effects of antidepressants also impairs LSD binding. V437A has similar effects, but S440A, a mutation that interferes with fluoxetine binding to TrkB, did not influence LSD binding (Fig. 1a). These findings indicate that the LSD binding site in the TrkB TMD partially overlaps with that of other antidepressants. LSD displays a high residence time at TrkB (koff = 0.0085 ± 0.0011 min−1), consistent with high affinity, but residence time is markedly reduced by Y433F (koff = 0.0240 ± 0.0037 min−1) (Extended Data Fig. 1c).
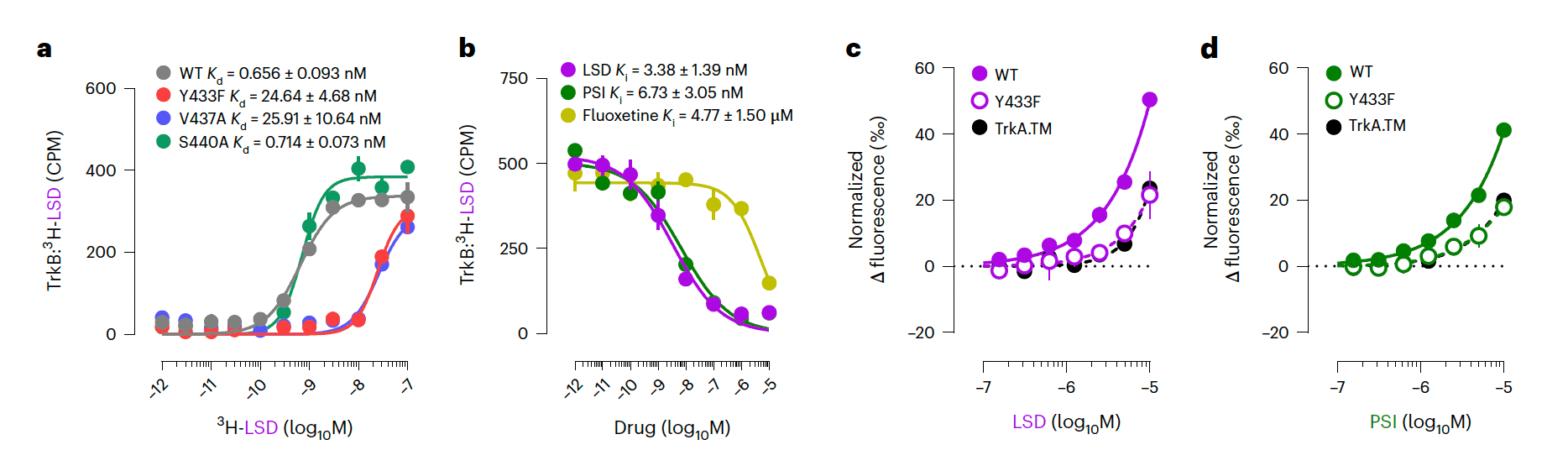
We also found that PSI displaces 3H-LSD bound to TrkB with high affinity (Ki = 6.73 ± 3.05 nM, Fig. 1b), while fluoxetine (Ki = 4.77 ± 1.50 µM, Fig. 1b) and ketamine (Ki = 20.82 ± 8.86 µM, Extended Data Fig. 1f) display substantially lower TrkB affinities. 2R,6R-hydroxynorketamine (R,R-HNK), an active metabolite of ketamine that binds to TrkB but shows low affinity to NMDA receptors, displaces LSD with high nanomolar concentrations (Ki = 166.00 ± 21.15 nM, Extended Data Fig. 1f). In turn, biotinylated R,R-HNK is displaced from TrkB by psychedelics at low nanomolar concentrations (Extended Data Fig. 1g). To further define the TrkB binding specificity, we tested displacement of 3H-LSD by other ergoline compounds. We found that lisuride, a nonpsychedelic ergoline 5-HT2A agonist, binds to TrkB at higher nanomolar concentrations in a linear manner, but other LSD-related compounds cabergoline and dihydroergotamine fail to displace 3H-LSD, as do the negative controls chlorpromazine and diazepam (Extended Data Fig. 1h,i). 5-HT2Aantagonists ketanserin and M100907 also fail to displace LSD binding to TrkB (Extended Data Fig. 1f,g). Importantly, no other known targets for psychedelics or antidepressants were identified by mass spectrometry (MS) in the samples used in our binding assays, confirming that LSD bound selectively to TrkB (Supplementary Table 2).
We confirmed binding to TrkB using microscale thermophoresis (MST) assay for unlabeled psychedelics to eGFP-tagged TrkB in lysates of HEK293T cells. MST confirmed that LSD, PSI and lisuride directly bind to native TrkB but not to Y433F or TrkA.TM mutants (Fig. 1c,d and Extended Data Fig. 1e). BDNF binding served as a positive control. The apparent potencies of all the compounds (including BDNF) were substantially lower when assayed with MST than the affinities obtained with binding assays, which may be due to interference by other lysate components.
Consistent with these observations, nuclear magnetic resonance (NMR) spectroscopy of isotope-labeled TrkB TMD incorporated into lipid bicelles detected chemical shift perturbations (CSPs) of amide groups of Y433, V437 and other residues within the TMD N-terminal side upon addition of LSD, which specifically take place in the dimeric state of the receptor (Fig. 2j,k and Extended Data Fig. 2). Together, these data suggest that the TrkB TMD dimer is a high-affinity primary target for psychedelics.
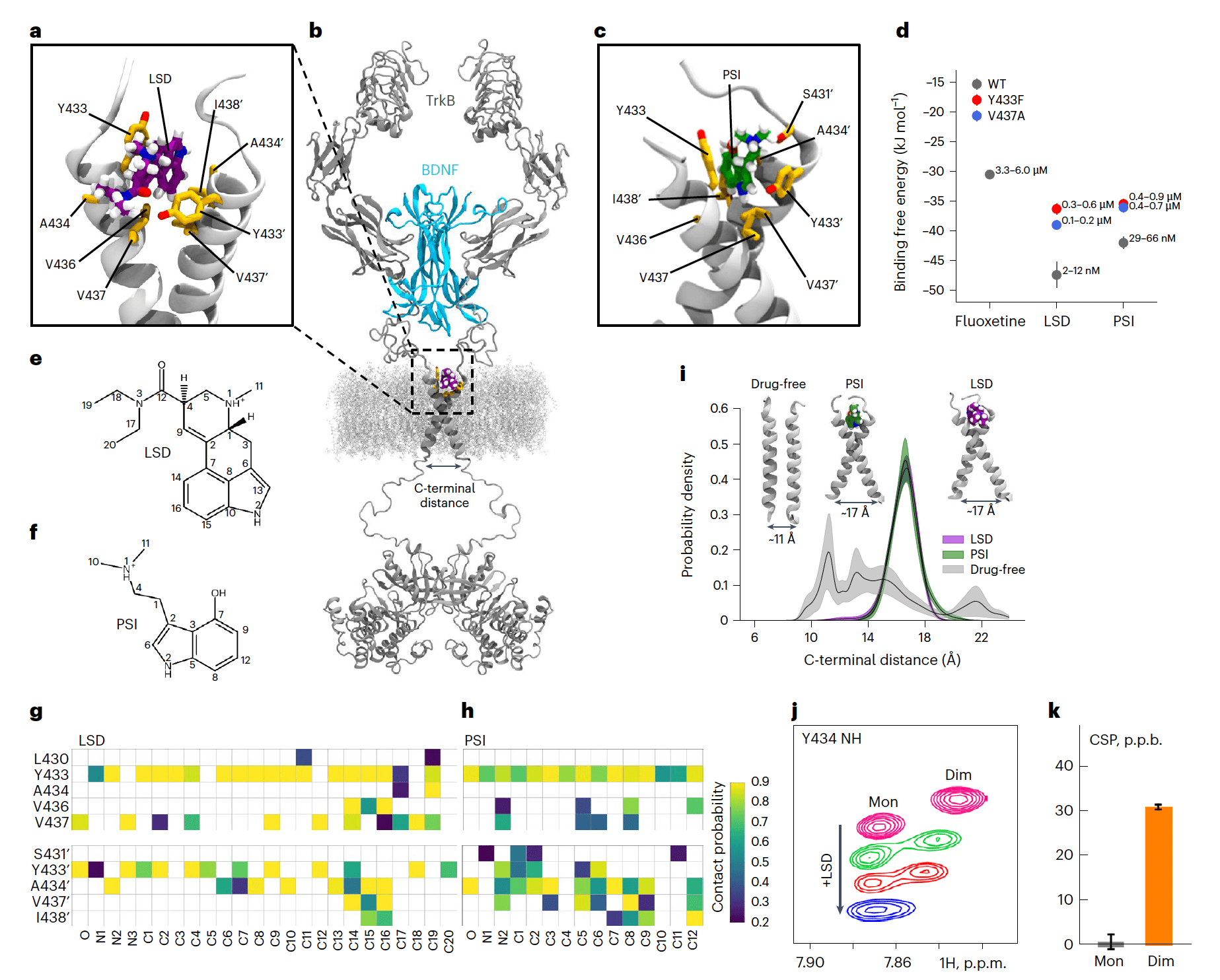
TrkB binding site of psychedelics
We used atomistic molecular dynamics (MD) simulations of the TrkB TMD dimers embedded in cholesterol (CHOL)-enriched membranes to identify the binding site of psychedelics. LSD and PSI spontaneously associate with TrkB TMD dimers. Consistent with our NMR structural data and mutagenesis binding studies, simulations revealed binding sites for LSD and PSI located in the extracellular-facing crevice of the crisscrossed TMD dimers such that both TMDs of a dimer are required for binding (Fig. 2a–c and Extended Data Fig. 3). Subsequent binding free energy estimations showed that LSD and PSI bind to the TMD of TrkB dimers with higher affinity than fluoxetine (Fig. 2d and Extended Data Fig. 4a). In agreement with our experimental data, Y433, A434 and V437 stabilize the binding of LSD and PSI (Fig. 1e–h and Extended Data Figs. 3 and 4b–d). Y433F and V437A mutations markedly decrease the binding affinity of psychedelics (Fig. 2d and Extended Data Fig. 4a). At the high CHOL concentration typical for synaptic membranes, LSD stabilizes the TrkB TMD dimer in a conformation where the distance between its Cα residues (L451–L453) is ~17 Å (Fig. 2i and Extended Data Fig. 4e). Thus, LSD stabilizes TrkB dimers in a conformation that is more favorable to activation by BDNF, akin to the mechanism previously identified for fluoxetine.
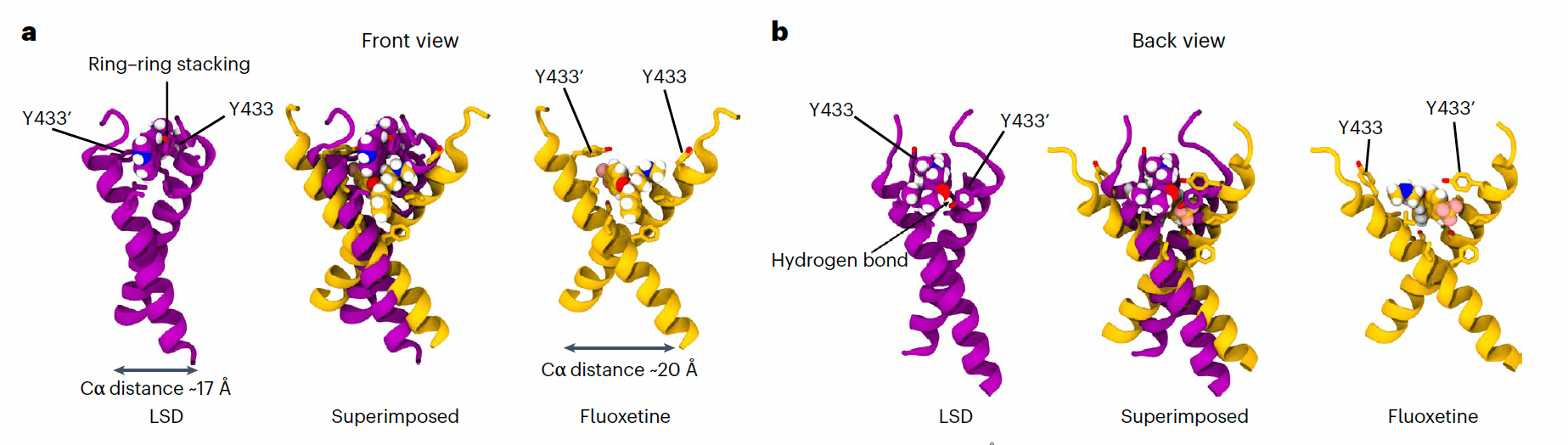
Notably, we found that the binding pockets for fluoxetine and LSD are distinct, even though their binding involves shared residues. Fluoxetine binds deeper within the TMD and requires membrane lipid stabilization, locking the TMD dimer in a more open cross-shaped conformation (Cα distance ~20 Å). In contrast, LSD binds closer to the membrane surface and establishes more stable interactions with the dimer: a hydrogen bond between the oxygen of LSD’s diethylamide group and the Y433 residue of one monomer, and pi-stacking of the ergoline aromatic backbone with the Y433 residue of the second monomer, locking TMD dimers in a tighter cross-shaped conformation (Cα distance ~17 Å) compared with fluoxetine (Fig. 3). This is consistent with the deeper-located mutation S440A interfering with fluoxetine but not with LSD binding (Fig. 1a). PSI displays a binding pocket, interacting residues and stable drug:TrkB complex conformation (Cα distance ~17 Å) similar to that of LSD (Fig. 2a–i and Extended Data Fig. 3). Atomistic simulations and free energy calculations show that lisuride binds to TrkB with reduced affinity when compared with LSD (Extended Data Fig. 4a), but interacting residues and TMD dimer conformation (Cα distance ~17 Å) of lisuride are similar to those of LSD and PSI (Extended Data Fig. 4f–i). In contrast, control compounds cabergoline and dihydroergotamine display very poor TrkB binding (Extended Data Fig. 4a). Thus, psychedelics and antidepressants have distinct binding pockets and stabilize different conformations of the TMD dimer, which may underlie the disparities observed among TrkB ligand affinities.
Psychedelics promote BDNF signaling
We used a split-luciferase complementation assay to investigate TrkB dimerization and interactions in response to psychedelics. LSD and PSI induce a fast and long-lasting increase in dimerization of wild-type (WT) TrkB at nanomolar concentrations, but this effect is abolished in WT:Y433F heterodimers (Y433F+/−) (Fig. 4a–c). Consistently, LSD increases phosphorylation of TrkB tyrosine 816 (pY816) residues in cortical and hippocampal cultures and in mouse brain samples after a single administration (Extended Data Fig. 5i,j,l,q), indicating facilitated dimerization. Lisuride similarly increases TrkB dimerization at higher concentrations (Extended Data Fig. 5a). Pretreatment with ketanserin or M100907 fails to prevent the effects of LSD and BDNF on TrkB dimerization in N2a cells that do not express 5-HT2A messenger RNA (Fig. 4d, Extended Data Fig. 5b,c and Extended Data Table 1), indicating that psychedelics induce TrkB dimerization in the absence of 5-HT2A and ketanserin and M100907 do not interfere with this effect.
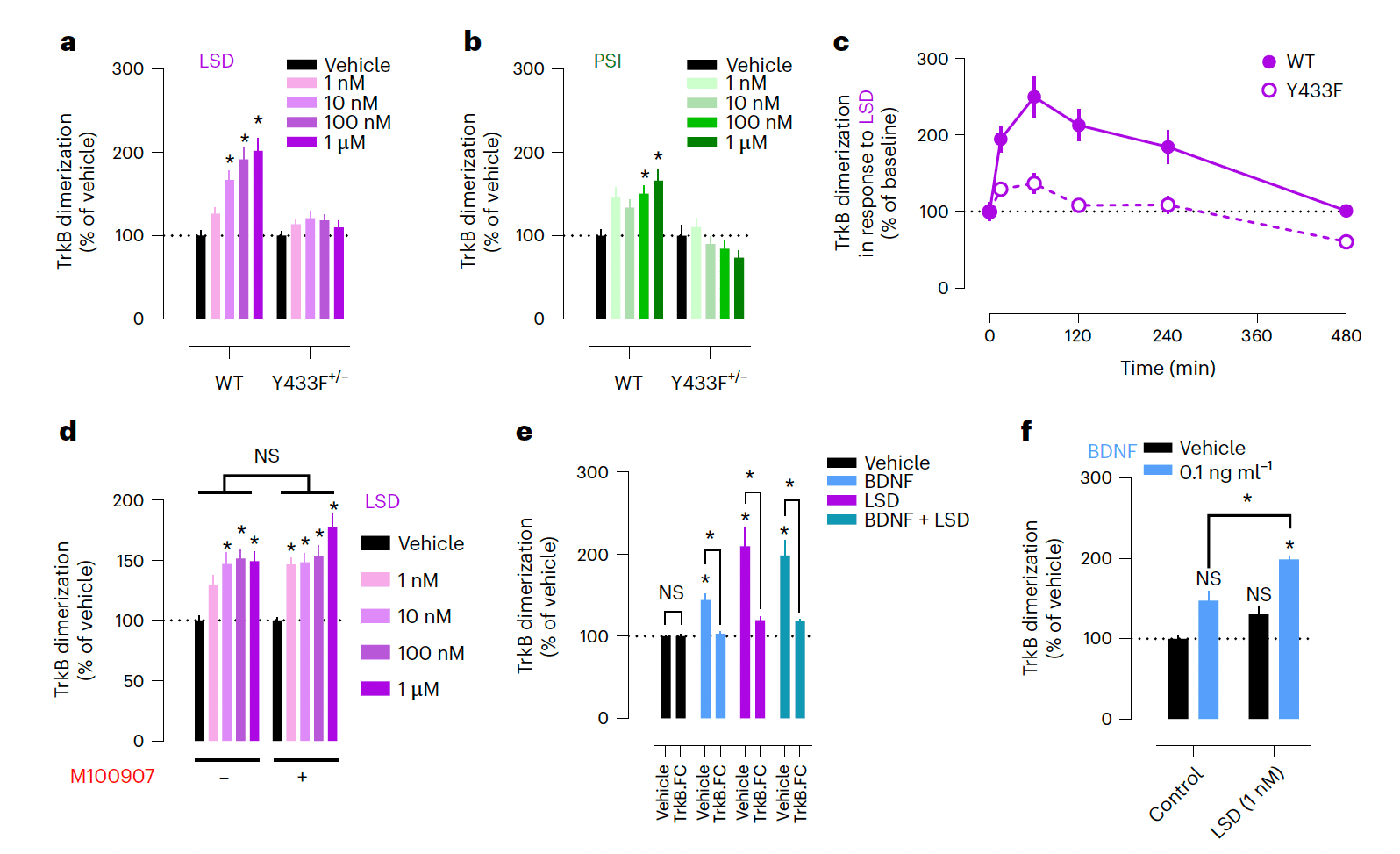
TrkB.FC, which sequesters extracellular BDNF, prevents the effects of LSD and BDNF on TrkB dimerization (Fig. 4e). Furthermore, LSD potentiates the effects of very low concentrations of BDNF (0.1 ng ml−1) on TrkB dimerization (Fig. 4f), presumably by stabilizing TrkB dimerization. No differences in total TrkB expression were observed between WT and Y433F+/− or following treatment with LSD or BDNF (Extended Data Fig. 5d). These data demonstrate that psychedelics do not directly activate TrkB, but their effects on TrkB dimerization are dependent on release of endogenous BDNF, consistent with an allosteric effect.
TrkB is mainly localized in intracellular vesicles and only transiently translocated to the cell surface where it is exposed to BDNF. Psychedelics rapidly increase the neuronal surface retention of TrkB, independently from 5-HT2A activation, and promote TrkB interaction with raft-restricted Src family kinase Fyn, indicating increased localization to raft-like synaptic membranes. This effect is lost in Y433F+/−heterodimers (Extended Data Fig. 5e–h).
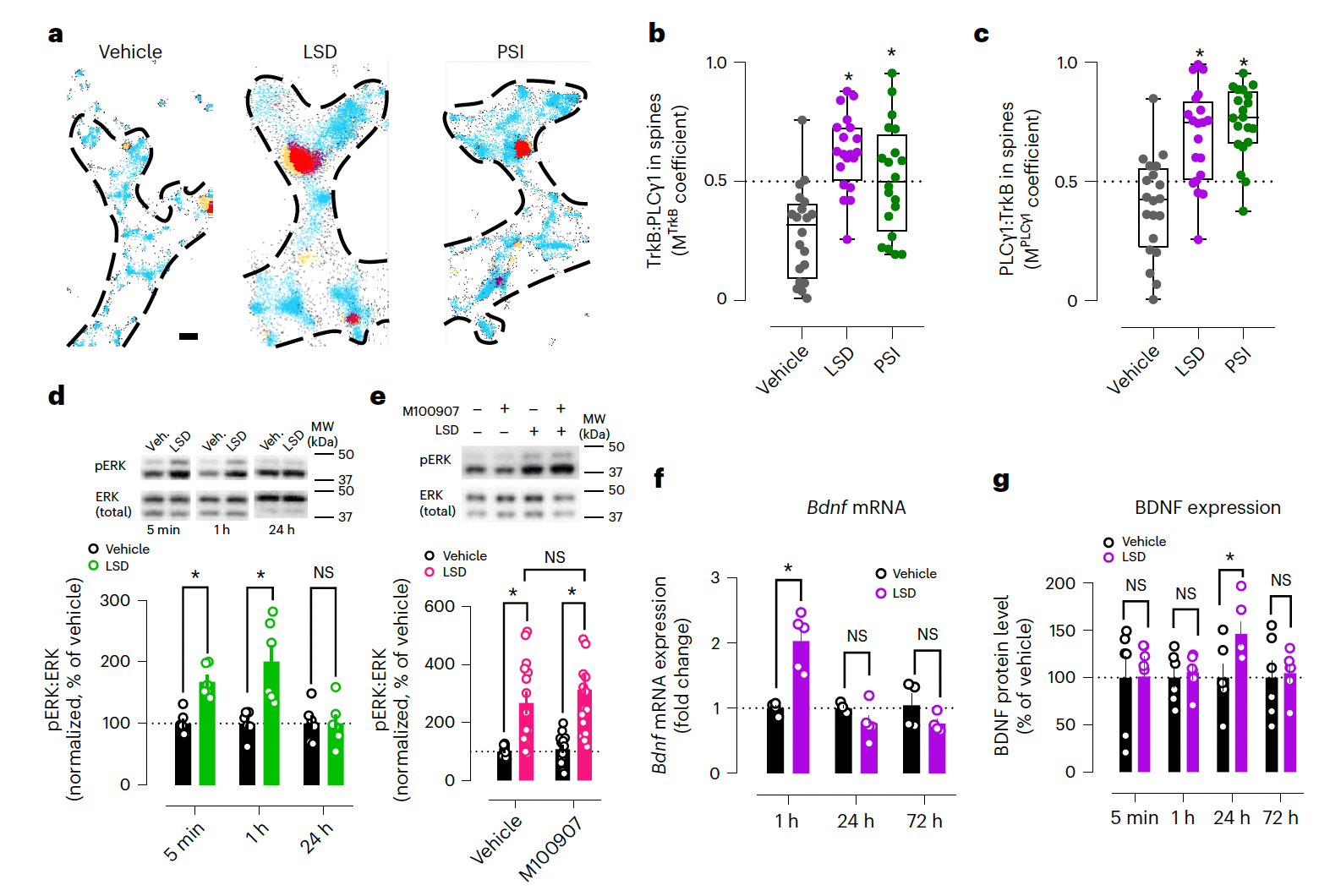
Psychedelics also promote BDNF downstream signaling. In neuronal cultures, LSD increases TrkB interaction with phospholipase C gamma 1 (PLCγ1) (Extended Data Fig. 5k), which docks on pY816 and regulates intracellular Ca2+ signaling and antidepressant action. TrkB interaction with PLCγ1 is also increased in the prefrontal cortex (PFC) and hippocampus of WT but not Y433F+/− mice, without affecting total TrkB expression (Extended Data Fig. 5m–p,r). Tracking single-molecule localizations of TrkB and PLCγ1 in neurons by direct stochastic optical reconstruction microscopy coupled with total internal reflection fluorescence (dSTORM/TIRF) confirmed that LSD and PSI increase TrkB:PLCγ1 colocalization in dendritic spines (Fig. 5a–c and Extended Data Fig. 5s–u). In addition, LSD rapidly increases phosphorylation of ERK (pERK), independently of 5-HT2A activation (Fig. 5d,e), and promotes phosphorylation of mTOR for at least 1 h (Extended Data Fig. 5v). Bdnf mRNA is upregulated at 1 h after LSD treatment and BDNF protein levels are elevated at 24 h (Fig. 5f,g) without any changes in Ntrk2 mRNA levels (Extended Data Fig. 5w). In summary, the effects of psychedelics on neurotrophic signaling are dependent on BDNF and TrkB, and do not require 5-HT2A activation.
Psychedelic-promoted plasticity depends on BDNF and TrkB
We used fluorescence recovery after photobleaching (FRAP) in neuronal cultures expressing GFP-tagged TrkB to investigate the effects of psychedelics on TrkB trafficking. FRAP revealed that psychedelics induce rapid TrkB trafficking into dendritic spines. LSD robustly increases fluorescence recovery (+42.42% increment, t1/2 = 13.53 s), followed closely by PSI (+22.72% increment, t1/2 = 13.99 s), when compared with vehicle treatment (Emax = 62.88%, t1/2 = 16.93 s). However, LSD (+4.80% increment, t1/2 = 16.00 s) and PSI (+8.63% increment, t1/2 = 15.18 s) failed to increase fluorescence recovery of Y433F compared with vehicle treatment (Emax = 70.82%, t1/2 = 11.08 s) (Fig. 6a,b and Extended Data Fig. 6a–d).
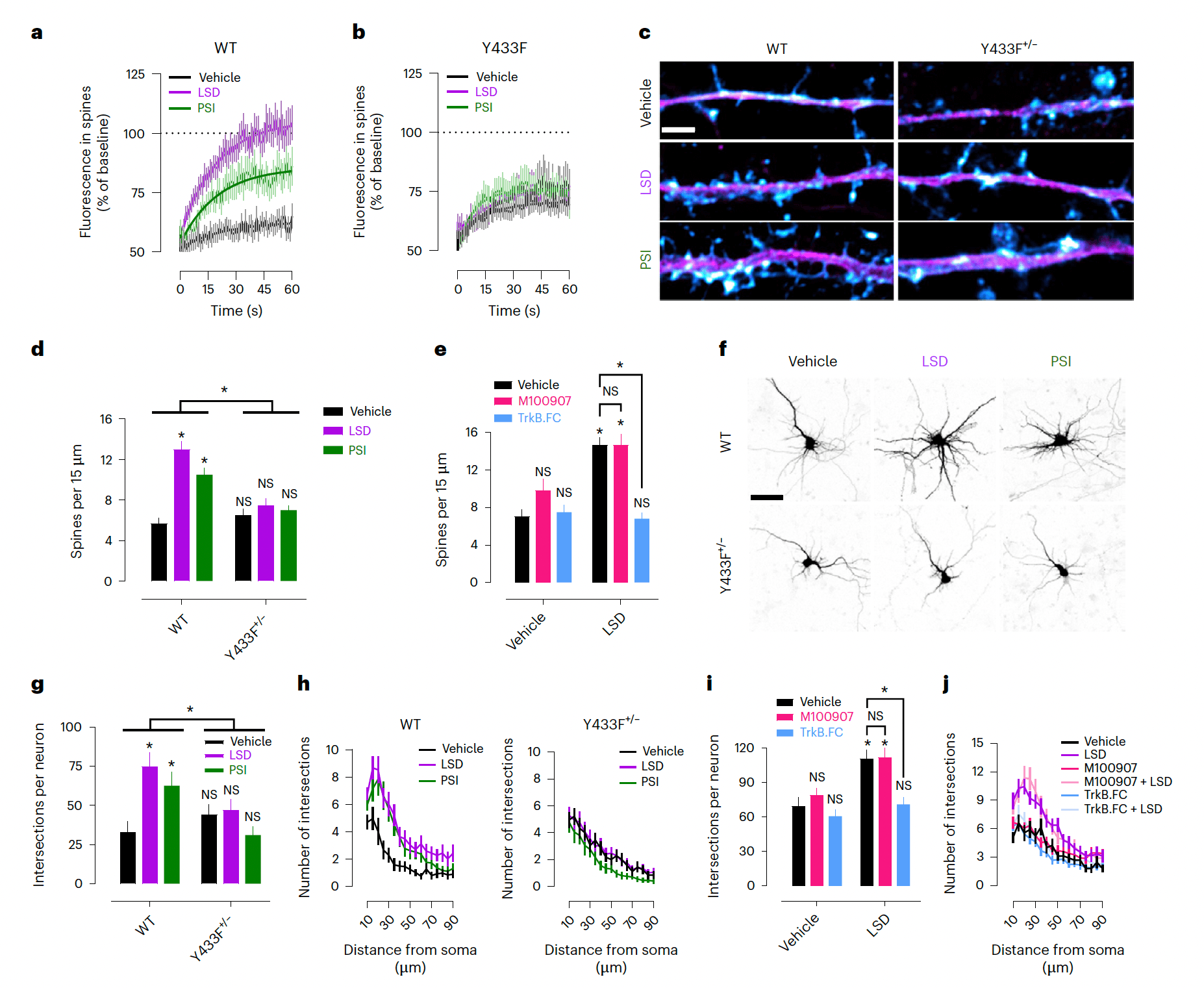
Previous findings have reported that psychedelics increase neurite outgrowth and spine formation in cultured neurons. Consistent with these findings, LSD and PSI increase spine density of mature neuronal cultures from WT mice at 24 h after treatment, but not of neuronal cultures from Y433F+/− mice. Notably, TrkB.FC but not M100907 prevented the spinogenic effect of LSD (Fig. 6c–e), indicating that this effect is dependent on BDNF release but not on 5-HT2A activation. Psychedelics also increase dendritic arbor complexity in neuronal cultures derived from WT but not Y433F+/− mice. Again, TrkB.FC but not M100907 prevented LSD effects on dendritogenesis (Fig. 6f–j). Ketanserin has previously been reported to prevent the effects of LSD on dendritic arbor complexity in cultured neurons; more research is needed to resolve this apparent discrepancy with present results, but the various off-target effects of ketanserin might at least partially explain this contradiction. Importantly, Y433F+/− neuronal cultures treated with BDNF showed increased dendritic arbor complexity and were indistinguishable from WT cultures (Extended Data Fig. 6e,f), indicating that the heterozygous Y433F+/− mutation does not interfere with the TrkB response to BDNF. Furthermore, Y433F+/− mice do not show any baseline differences from WT TrkB mice, and Y433F+/− neuronal cultures and mice respond normally to BDNF in plasticity-related and behavioral assays. These results indicate that Y433F+/− does not compromise TrkB function or BDNF responses; it only abrogates the binding of psychedelics and antidepressants to TrkB and their effects on plasticity. Together these findings are consistent with previous reports of TrkB involvement in psychedelic drug-induced plasticity, and support the notion that psychedelics are not direct TrkB agonists, but they facilitate the effects of endogenous BDNF, independently of 5-HT2A activation.
LSD effects on network plasticity and behavior
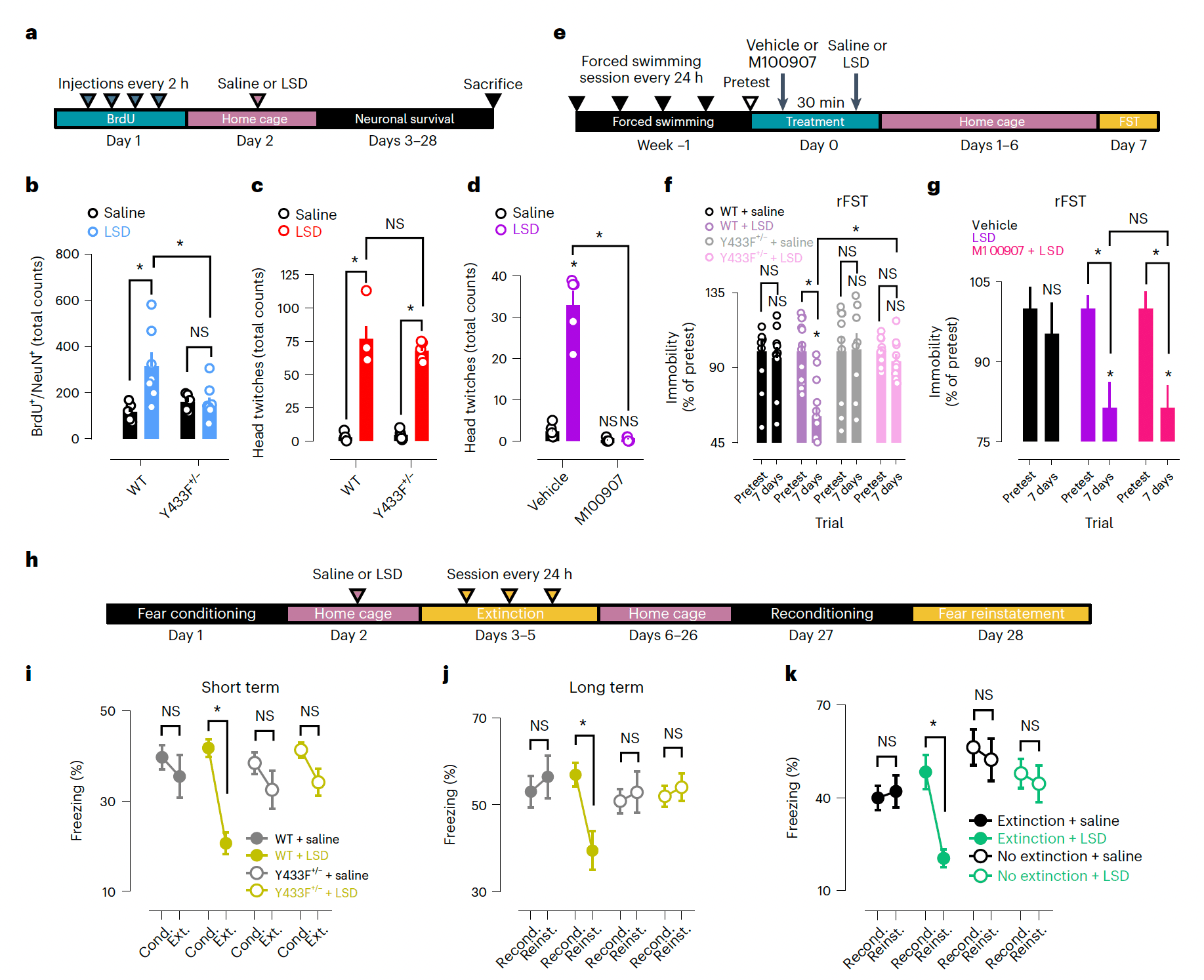
Chronic treatment with conventional antidepressants or a single administration of ketamine enhance neurogenesis and long-term neuronal survival of dentate granule cells (DGCs) of the hippocampus, and these effects are dependent on BDNF signaling through TrkB. We found that LSD doubles the number of surviving DGCs of WT mice, but not of Y433F+/− mice, at 4 weeks after a single administration (Fig. 7a,b and Extended Data Fig. 6g,h), indicating that the effects of LSD on DGC survival are mediated by their binding to TrkB.
We and others have previously shown that antidepressants and ketamine reactivate critical period-like plasticity in the adult visual cortex. We found that LSD also promotes visual plasticity and facilitates a shift in ocular dominance (OD) in favor of the open eye in the primary visual cortex of mice following a week of monocular deprivation (Extended Data Fig. 7a,b).
The head-twitch response has been used in rodents as a reporter of the hallucinogenic effects of psychedelics in humans and this response is known to be mediated by 5-HT2A receptors. We found that the head-twitch response occurs normally in Y433F+/− mice treated with LSD, but is blocked by M100907 (Fig. 7c,dand Extended Data Fig. 7c,d). These results suggest that, as far as the head-twitch response reliably reports hallucinogenic actions of LSD in humans, these effects are dependent on 5-HT2A activation but detached from TrkB activity, which indicates a dissociation between the hallucinogenic and plasticity-promoting effects of psychedelics.
In a rodent model of chronic stress consisting of repeated sessions of the forced swimming test (rFST), LSD produced a sustained antidepressant-like effect 7 days after a single administration in WT but not Y433F+/− mice. Pretreatment with M100907 did not prevent the antidepressant-like effect of LSD (Fig. 7e–g). LSD also facilitates contextual extinction of conditioned fear response 72 h after a single administration, which persists for at least 4 weeks. However, these short- and long-term effects are lost in Y433F+/− mice. Importantly, LSD alone does not bring about fear extinction, as extinction training is required to produce a sustained decrease in behavioral freezing after LSD treatment (Fig. 7h–k and Extended Data Fig. 7e–s). This is reminiscent of the effects of other antidepressants that depend on environmental input to exert their therapeutic-like effects, and are consistent with the importance of ‘set and setting’ in modulating psychedelic effects in humans. Taken together, these results strongly suggest that TrkB mediates the plasticity-related and antidepressant-like effects of LSD at the network and behavioral levels but is not involved in its hallucinogenic-like action.
Discussion
Recent clinical studies suggesting that psychedelics produce rapid and long-lasting antidepressant effects have received tremendous attention. Mystical experiences induced by psychedelics have been associated with their clinical efficacy. However, the hallucinogenic effects of psychedelics limit their widespread clinical application, as their administration is restricted to clinical settings that often require intensive monitoring. A number of recent observations suggest that the antidepressant and plasticity-promoting effects of psychedelics may be dissociable from their hallucinogenic effects. First, plasticity-like effects of psilocybin are not blocked by the 5-HT2A antagonist ketanserin in mice, while it blocks the head-twitch response, a 5-HT2A-mediated behavior in rodents used as a proxy for hallucinogenic action of psychedelics in humans. Second, derivatives of psychedelic compounds have recently been introduced that promote plasticity and antidepressant-like behavior but do not seem to produce head-twitch responses. However, detailed information concerning the sites that mediate these differential actions have remained unclear.
Here we show that psychedelics promote neuroplasticity and plasticity-related behavioral effects through their high-affinity binding to TrkB (Extended Data Fig. 8). Psychedelics are not direct TrkB agonists, as extracellular BDNF is necessary for their effects on TrkB dimerization and plasticity, but they act allosterically by facilitating the effects of endogenous BDNF released in active synapses, similar to what we recently found for other antidepressants. Activity-dependent release of BDNF in stimulated synapses helps to selectively stabilize active synapses at the expense of inactive ones, which is critical for Hebbian-type plasticity. Direct TrkB agonists are expected to indiscriminately activate TrkB in active and inactive synapses, which leads to gradual reduction of signal-to-noise ratio within neuronal networks. Therefore, through a positive allosteric modulation of BDNF signaling, psychedelics selectively promote, maintain and strengthen activity-dependent plasticity in active synapses.
A point mutation in the critical Y433 residue that impairs psychedelics binding to TrkB also abolishes induction of neuroplasticity and long-term plastic responses, but does not affect the head-twitch response. Moreover, 5-HT2A antagonists fail to prevent psychedelic-induced TrkB dimerization and neurotrophic signaling, spinogenesis, dendritogenesis and antidepressant-like behavioral effects. These findings are perhaps not completely surprising, as there is still some debate in the field about whether 5-HT2A receptors mediate the therapeutic effects of psychedelics. Overall, our results suggest that the TrkB-dependent effects of psychedelics on plasticity can be detached from their hallucinogenic-like effects mediated by 5-HT2A. We further show that lisuride also binds to TrkB, albeit with a lower affinity than LSD and PSI, which is consistent with recent evidence suggesting that lisuride increases dendritogenesis and spinogenesis in culture neurons.
We have recently found that a dimer of TrkB TMDs contains a binding site for antidepressants, and that their plasticity-promoting and antidepressant-like effects are dependent on this interaction. However, the affinity of conventional antidepressants to TrkB, albeit pharmacologically relevant, is low. Brain concentrations required for selective serotonin reuptake inhibitors binding to TrkB are achieved only after long-term treatment, which may partially explain the delay in the onset of their antidepressant action. Psychedelics readily penetrate into the brain after a single dose and, as shown here, bind to TrkB with much higher affinities than antidepressants in current clinical use. This may contribute to the fast and potent induction of neuroplasticity and more persistent behavioral effects produced by psychedelics when compared with other antidepressants.
We have here characterized at an atomistic level of detail the binding sites of LSD, PSI and lisuride at TrkB TMD dimers, and used several control compounds to define binding specificity. We have also described the TrkB TMD conformational changes induced by psychedelics. Notably, the TMD binding pockets and TrkB conformational changes induced by antidepressants and psychedelics are distinct. However, it is clear that these compounds have different polypharmacological profiles beyond TrkB and 5-HT2A that probably contribute to the overall effects and timeframes of each particular drug.In conclusion, our findings support TrkB as the key target for psychedelic drug-induced plasticity. These data confirm TrkB as a common binding target for antidepressants and open an avenue for structure-based design of high-affinity TrkB-selective ligands with fast and long-lasting antidepressant action, but potentially devoid of hallucinogenic-like activity.