Abstract
Post-traumatic stress disorder (PTSD) is a maladaptive and debilitating psychiatric disorder, characterized by re-experiencing, avoidance, negative emotions and thoughts, and hyperarousal in the months and years following exposure to severe trauma. PTSD has a prevalence of approximately 6–8% in the general population, although this can increase to 25% among groups who have experienced severe psychological trauma, such as combat veterans, refugees and victims of assault. The risk of developing PTSD in the aftermath of severe trauma is determined by multiple factors, including genetics — at least 30–40% of the risk of PTSD is heritable — and past history, for example, prior adult and childhood trauma. Many of the primary symptoms of PTSD, including hyperarousal and sleep dysregulation, are increasingly understood through translational neuroscience. In addition, a large amount of evidence suggests that PTSD can be viewed, at least in part, as a disorder that involves dysregulation of normal fear processes. The neural circuitry underlying fear and threat-related behaviour and learning in mammals, including the amygdala–hippocampus–medial prefrontal cortex circuit, is among the most well-understood in behavioural neuroscience. Furthermore, the study of threat-responding and its underlying circuitry has led to rapid progress in understanding learning and memory processes. By combining molecular–genetic approaches with a translational, mechanistic knowledge of fear circuitry, transformational advances in the conceptual framework, diagnosis and treatment of PTSD are possible. In this Review, we describe the clinical features and current treatments for PTSD, examine the neurobiology of symptom domains, highlight genomic advances and discuss translational approaches to understanding mechanisms and identifying new treatments and interventions for this devastating syndrome.
Post-traumatic stress disorder (PTSD) is a severe psychiatric disorder that develops in the months and years following exposure to severe trauma1. The characteristic symptoms of the disorder include re-experiencing of trauma memories, avoidance of cues that remind the individual of the trauma, negative emotions and thoughts, and hyperarousal. PTSD has a prevalence of approximately 6% in the general population but can occur in 25–35% of individuals who have experienced severe trauma, for example, combat veterans, refugees and victims of assault. The risk of developing PTSD after trauma is multi-factorial, and involves genes and the environment. Although at least 30–40% of this risk is heritable, it is also influenced by past personal history, including prior adult and childhood trauma, and psychological factors that might differentially mediate the regulation of fear and emotion.
Hyperarousal. A core feature of post-traumatic stress disorder (PTSD) that includes irritability, panic and disruptions in sleep and cognitive function. |
Two of the more well-known factors that influence the risk of PTSD are the type of trauma and the sex of the individual. Although some studies suggest that the symptoms and biological mechanisms of PTSD are similar across different types of trauma and different degrees of exposure, some clear differences have been reported. Notably, among the various types of trauma, childhood trauma and interpersonal assault and violence seem to carry the greatest risk of subsequent development of PTSD. Furthermore, biological findings suggest that military and civilian trauma exposures might involve different mechanisms and have different biomarkers of PTSD risk. However, teasing apart the influence of trauma type from that of other components that often make up cohort differences (for example, sex, age, premorbid functioning, social support and other risk or resilience factors) remains difficult. One of the most important findings related to the risk of PTSD is the approximately 2:1 ratio of increased PTSD prevalence in women compared with men. This difference in risk is likely to be influenced by differences in the types of trauma experienced by individuals of different sexes as well as by differences in biology, for example, the regulation of risk and resilience responses by sex hormones.
Because the aetiology of PTSD stems from a specific, highly traumatizing, fear-evoking experience (often called the ‘index trauma’), it is considered a prototypical example of a psychiatric disorder that can be better understood by modelling the interaction of environmental influences with genetic vulnerability. In addition, considerable evidence supports a conceptual framework in which PTSD can be viewed, at least in part, as a disorder of fear dysregulation, which offers opportunities to advance the field through translational neuroscience approaches. The neural circuitry underlying fear behaviour in mammals, including the circuit that connects the amygdala, hippocampus and medial prefrontal cortex, is among the most well understood in neuroscience. In addition, the study of fear-related and threat-related behaviour and its underlying circuitry has led to some of the most rapid advances in our understanding of learning and memory processes. By combining molecular–genetic approaches with a mechanistic understanding of fear circuitry, we believe great progress in the understanding, diagnosis, and treatment of PTSD is imminent.
In contrast to the promise and progress of current scientific approaches, treatment options for PTSD in the clinical setting remain limited. The best currently available treatment for PTSD is exposure-based, trauma-focused cognitive behavioural therapy, which is thought to act via modulation of the neurocircuitry of fear extinction. No psychotropic medications have been developed and approved specifically for PTSD. Instead, the only FDA-approved treatments for the disorder are two antidepressant medications: sertraline and paroxetine. Considering that these medications often fail to address the full range of PTSD symptoms, a better mechanistic understanding of the pathogenesis and biology underlying intermediate phenotypes of the condition is urgently needed in order to accelerate the identification of novel targets for improved treatments. In this Review, we first describe the clinical features of and current treatments for PTSD. We then go on to discuss neuroanatomical and molecular–genetic approaches to the study of PTSD and relate them to a translational understanding of fear circuitry, with the aim of exploring possible advances in the conceptual framework, diagnosis and treatment of PTSD.
Startle response. A reflex that occurs rapidly and unconsciously in response to an external stimulus such as a noise burst. |
|---|
Hypervigilance. A core feature of post-traumatic stress disorder (PTSD) characterized by a heightened state of active threat assessment. |
Clinical features of PTSD
To meet the diagnostic criteria for PTSD outlined in the Diagnostic and Statistical Manual of Mental Disorders fifth edition (DSM-5), an individual must first have had a traumatic experience that involved being exposed to actual or threatened death, serious injury, or sexual assault. Individuals who have symptoms of PTSD during the first month following trauma exposure are considered to have ‘acute stress disorder’ as, in many such individuals, the symptoms naturally resolve in the days and weeks following the initial shock and emotional upheaval of the traumatic exposure. Individuals with symptoms of PTSD that are consistent for at least 2 weeks and are ongoing at least 1 month after trauma exposure are considered to have a diagnosis of PTSD.
PTSD is characterized by four symptom clusters: intrusion and re-experiencing; avoidance and numbing; negative mood and impaired cognition; and hyperarousal. Intrusion and re-experiencing symptoms comprise DSM-5 criterion B and include unwanted intrusive memories ranging from mild unwanted memories to full dissociative flashbacks during which the individual momentarily believes they are re-living the traumatic experience. This symptom cluster also includes disturbing, and at times overwhelming, nightmares of the traumatic event. DSM-5 criterion C includes avoidance of any reminder cues, contexts or people related to the trauma, which can become a substantial source of disability as individuals become more isolated, often not leaving their home owing to the generalization of triggering experiences. ‘Negative alterations in cognition and mood’ is a broad cluster of symptoms that comprises DSM-5 criterion D and includes trauma-related depressive-like symptoms, anhedonia, emotional numbing and problems concentrating. Hyperarousal symptoms constitute DSM-5 criterion E and include decreased sleep, increased startle response, hypervigilance and irritability, as well as aggressive and arousal-related self-destructive behaviour.
In 2013, a new dissociative subtype of PTSD was added to DSM-5 with the aim of improving the characterization of individuals with PTSD who also experience pervasive dissociative symptoms. To meet criteria for the dissociative subtype, an individual must meet full criteria for PTSD while also experiencing substantial symptoms of depersonalization and/or de-realization. The DSM-5 defines depersonalization as “experiences of unreality, detachment, or being an outside observer with respect to one’s thoughts, feelings, sensations, body, or actions” and de-realization as “experiences of unreality or detachment with respect to surroundings”. This dissociative subtype was added in recognition that a subset of individuals with PTSD and dissociative symptoms can be reliably identified in both military and civilian samples. Neurobiological and clinical research in PTSD also supports the existence of a dissociative subtype, as further outlined below.
Studying PTSD: reasons for optimism
Often, PTSD is viewed as a psychiatric syndrome that could be particularly tractable. Several reasons exist to support this optimistic view. First, there is a high level of intersection between the clinical symptoms of PTSD and our existing knowledge of the underlying neurocircuitry (BOX 1). Moreover, threat-related behaviours and their underlying neural circuitry are highly conserved across mammals, including from mice to humans. Decades of work investigating the neurobiology of fear and threat behaviours in animal models can thus be leveraged to advance our understanding of the dysregulation of these systems in individuals with PTSD. Second, PTSD is among the few psychiatric syndromes for which the timing and cause of onset (that is, the aetiology) of the illness — exposure to the index trauma — is understood. Although much of the research into PTSD focuses on identifying why some people who are exposed to trauma go on to develop the disorder and others are resilient, in all cases, trauma exposure is required for PTSD development. Indeed, much of the work on trauma exposure has provided new insights into mechanisms of resilience. Studies have shown that resilience can be genetically heritable and that common polymorphisms contribute to resilience after trauma exposure. Furthermore, studies in at-risk populations have examined different psychological coping styles and brain activity patterns that support resilience as well as the effects of resilience in buffering against substance use disorders and other negative sequelae of trauma exposure.
Box 1 |. Improving alignment of psychiatry and neuroscience.
Rapid advances in technology are changing the ways in which illnesses such as post-traumatic stress disorder (PTSD) are diagnosed, treated and studied. In humans, these advances consist of continued refinements in brain imaging, increasing use of smart devices and wearables, and dramatic advances in the efficiency of genetic analyses. In animal models, a corresponding evolution of precision molecular techniques to probe and dissect neural circuitry is ongoing. Despite these advances, psychiatry and basic neuroscience continue to evolve largely in parallel, with few examples of the fields aligning and integrating to produce transformative changes in human health. Although many factors contribute to these gaps, most are related to a lack of forward (animal to human) and back translation (human to animal) of key discoveries, leading to questions about the utility of model systems in drug development.
Paths forward
Key attributes for experimental approaches that will enable transformational advances in research on PTSD and other psychiatric illnesses:
Translational relevance: the use of similar or identical end points in humans and laboratory animals.
Continuous data collection: end points that are measured over long periods (hours, days or weeks).
Objectiveness: data collected and analysed using rigorous algorithms, involving minimal handling or visual scoring.
Together, the above attributes make experimental findings more robust, reproducible and predictive of effects in other species. Many tools for the collection of data that have these key attributes are now available and include digital devices, such as smartphones and activity trackers, for use in humans and machine learning-based behavioural analysis in animals.
Translationally aligned end points
The following are representative examples of end points that fulfil the three key attributes above and are dysregulated in PTSD and by stress exposure in animal models:
Behavioural
Acoustic startle
Sleep
Diurnal fluctuations in motor activity rhythms
Diurnal fluctuations in core body temperature rhythms
Attention
Cognitive control
Reward learning
Biomarkers
Blood based
Peripheral samples and biopsies
Post-mortem analyses
Note that some techniques, such as brain imaging, fulfil the three key attributes but often involve substantial procedural deviations (for example, restraint or anaesthetic) or fundamental differences in capabilities (for example, ability to understand instructions, guidance or reassurance) across species.
Thus, we can study the onset of PTSD in the immediate and prolonged aftermath of trauma in ways that are not possible for other neuropsychiatric disorders, raising the potential for primary and secondary prevention of PTSD development based on knowledge of the processes of trauma memory formation, sensitization and generalization over time. The mechanisms of trauma memory encoding and consolidation as well as those of extinction memory formation, discrimination versus generalization of fear, and other emotional memory processes (for example, reconsolidation), all rely upon synaptic plasticity and systems memory processing. Ongoing research into biomarkers, including those that could be detected in the blood or other tissue samples, is bringing the field closer to the possibility of meaningful PTSD prevention. Furthermore, numerous translational studies have identified biological systems and molecular pathways that could be targeted to buffer trauma memory consolidation in the Emergency Department or the battlefield; pilot prevention studies have been performed but none have yet been definitive. The field of neuroscience has made tremendous progress towards understanding mechanisms of fear memory formation and regulation over the last decades; this progress has direct implications for our understanding of trauma memories and avenues for therapeutic intervention in PTSD.
Classical conditioning in PTSD
The neurobiology of Pavlovian threat memory acquisition is well characterized. This process is particularly relevant for understanding PTSD as the PTSD-inducing trauma exposure is frequently considered to be an example of human naturalistic fear conditioning. In experimental paradigms for assessing threat memory, a neutral stimulus (for example, a light, tone or smell) is presented repeatedly in tandem with an aversive stimulus (unconditioned stimulus; for example, a shock). After these repeated combined presentations, the individual (person or laboratory animal) learns that the previously neutral stimulus predicts the aversive unconditioned stimulus, transforming it into a conditioned stimulus. Consequently, the individual will exhibit fear-related behaviour in response to the conditioned stimulus, regardless of whether or not it is accompanied by the aversive unconditioned stimulus. Evidence from neuroimaging, lesion and pharmacology studies across species suggests that information about the conditioned stimulus and the unconditioned stimulus converge at the lateral and basolateral amygdala via afferents from the thalamus and cortex. Pairings of the conditioned stimulus and unconditioned stimulus induce synaptic plasticity at the level of the basolateral amygdala. Subsequent activation of the central amygdala, via input from the basolateral amygdala, elicits conditioned stimulus-elicited fear responses, including freezing, increased heart rate and potentiated startle, by activating downstream brain areas like the hypothalamus, locus coeruleus and other brainstem nuclei.
Cre-recombinase-dependent expression. A method of inducing alterations in gene expression involving the ability of the enzyme Cre-recombinase to induce site-specific recombination of genetic material. |
By contrast, extinction learning — during which fear is diminished through the process of exposure to the fear-eliciting conditioned stimulus in the absence of the aversive unconditioned stimulus — is conceptualized as a process that involves new learning that occurs through multiple mechanisms and suppresses, rather than erases, existing aversive memories. Dynamic changes in molecular mechanisms controlling GABAergic activity have been observed during fear acquisition and extinction learning in rodents (laboratory rats, mice); the observed changes suggest that an increase in amygdala GABAergic transmission has a role in extinction. Furthermore, in vivo electrophysiology studies identified a subset of excitatory projection neurons in the basolateral amygdala that exhibit increased firing rates during extinction. Note that modern genetic approaches to understanding the neural circuits of behaviour in animal models, including optogenetic, chemogenetic and cell type-specific manipulations (BOX 2), are revolutionizing our mechanistic understanding of circuits and behaviours. Using these tools, researchers have identified ‘extinction neurons’ that are responsive to the conditioned stimulus during extinction trials but not during fear conditioning, and seem to actively suppress the prior fear memory when in a context associated with safety. Additionally, in a study in rats, infralimbic cortical neurons exhibited increased conditioned stimulus-elicited firing during extinction retention and recall compared with baseline, suggesting that activity of the infralimbic cortex is crucial for inhibition of fear. Reminders (or retraining) were able to restore the original threat response more quickly than the original training regimen, suggesting that the memories were suppressed as opposed to erased.
Box 2 |. Experimental tools for dissection of threat circuits in animal models.
Optogenetics
Optogenetics enables spatiotemporally precise optical control of specific neuronal populations. To manipulate neuronal firing, the light-sensitive protein channelrhodopsin 2 (ChR2) is expressed in targeted, neurochemically and functionally identifiable neurons using viral delivery systems or genetic interventions and then activated by light delivered at standardized frequencies through an implanted optical fibre. ChR2 is a non-selective cation-permeable ion channel activated by blue light, resulting in membrane depolarization and the triggering of spike firing. Neurons can also be silenced with the light-sensitive inhibitory chloride pump halorhodopsin or firing can be inhibited by archaerhodopsin, a proton pump activated by yellow light. The use of viral vectors that transfect neighbouring neurons in anterograde-preferring or retrograde-preferring directions can achieve projection-specific targeting of these light-sensitive proteins, enabling the manipulation of behaviour-controlling pathways. The addition of Cre-recombinase-dependent expression of genes in specific cell types further enhances the specificity of circuit-level activity manipulations.
Chemogenetics
Although optogenetics is commonly used for temporally precise manipulations, chemogenetics enables the control of naturally occurring neuronal firing patterns over extended periods of time. Chemogenetics uses the expression of DREADDs (designer receptors exclusively activated by designer drugs) in transgenic mice or via viral vectors. DREADDs can be activated by ‘designer’ ligands (for example, clozapine N-oxide (CNO)) that do not have natural targets in the brain and are administered systemically or into discrete brain regions. Several DREADDs exist but hM4Di (derived from the M4 muscarinic receptor linked to the Giprotein) is often used for inhibition, whereas a Gq-coupled M3 muscarinic receptor-based DREADD (hM3Dq) is used for activation. Similar to optogenetics, DREADDs can be expressed in a cell type-specific and circuit-specific manner. After systemic injection of CNO, the natural firing patterns of DREADD-expressing neurons are activated or inhibited for prolonged periods of time (up to hours), facilitating the understanding of circuits and complex behaviour.
Activity-dependent imaging
Activity-dependent imaging relies on genetically encoded indicators of neuronal and network-level activity, including indicators of vesicular release, neurotransmitters, voltage and calcium. GCaMP is a widely used genetically encoded calcium indicator that becomes fluorescent when bound to Ca2+. Detecting Ca2+-dependent GCaMP fluorescence is possible with fibre photometry, in which a small fiberoptic tube or wire is embedded next to GCaMP-expressing neurons in specific brain regions to detect cell type-specific population activity. Separately, mini-microscopes can be implanted in brain regions to determine real-time genetically dependent cell activity. The use of activity-dependent indicators enables the estimation of neuronal activity in specific brain regions and specific neuronal subtypes at different stages of behaviour. Combining cell type-specific and projection-specific activity manipulations with real-time imaging of neuronal activity provides unprecedented insights into the function of behaviour-controlling microcircuits.
Overall, studies performed in rodent model systems and humans since the 1980s and 1990s have repeatedly indicated that Pavlovian threat conditioning occurs in part via amygdala circuits that activate downstream ‘reflexive’ threat responses. These systems seem to go awry, either through ‘over-learning’ at the time and in the aftermath of the initial trauma exposure, or through the inability to normally recover (via extinction) healthy safety learning following trauma. Studies in the laboratory setting have shown that individuals with PTSD have increased fear conditioning, deficits in extinction, and increased physiological (for example, sympathetic responses measured with galvanic skin response) and brain correlates of fear-responding (for example, amygdala and anterior cingulate hyperarousal) when compared with healthy control participants (BOX 1).
Neuroanatomy of PTSD
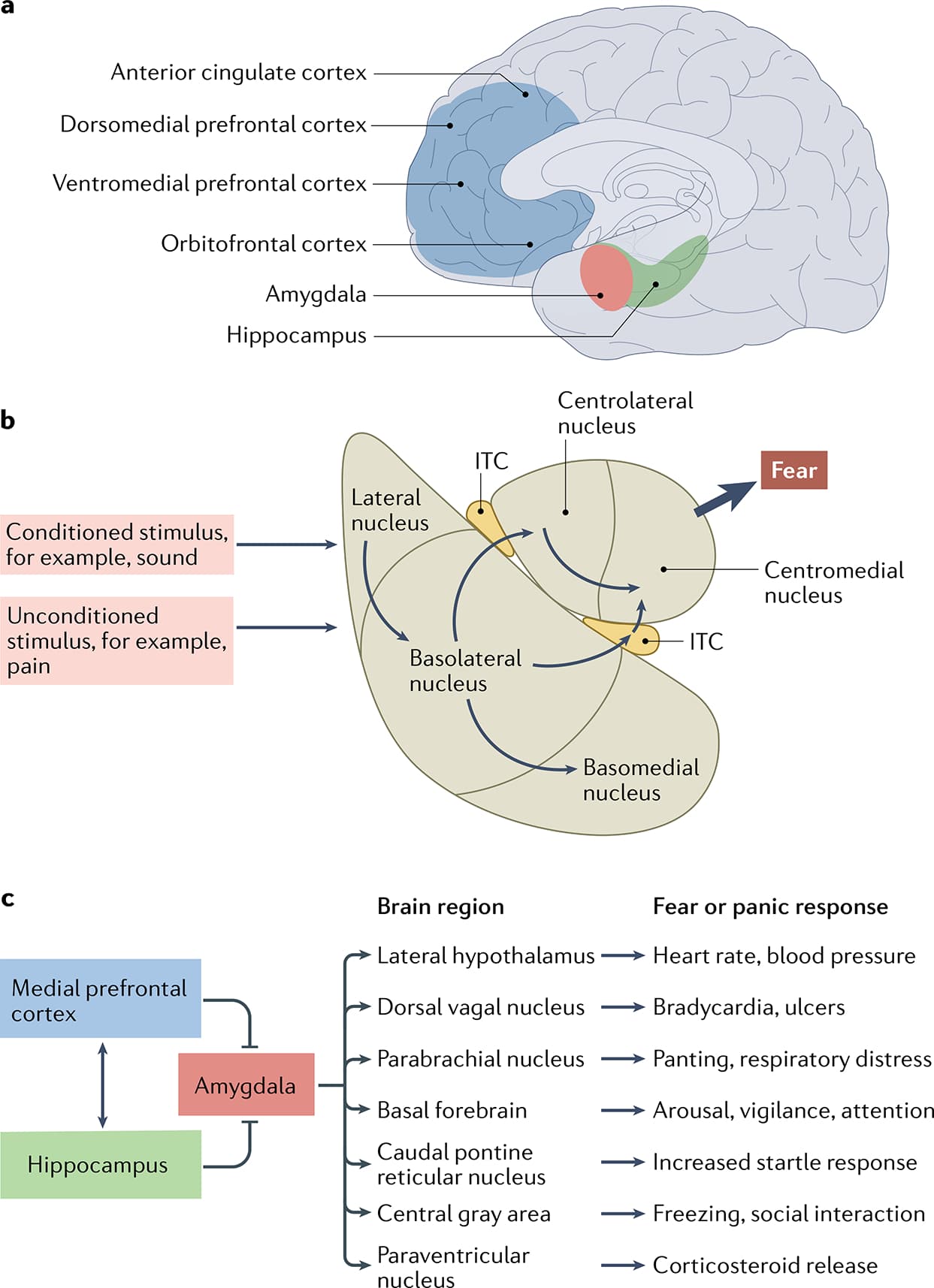
The brain regions most consistently associated with PTSD include the amygdala complex, hippocampus, insular cortex and areas of the prefrontal cortex, including the subgenual and dorsal anterior cingulate (FIG. 1a). Although they do not receive as much attention, the dorsolateral prefrontal cortex, striatum, thalamus and sensory areas are also likely to be involved. These brain regions work in concert for the initial acquisition and later expression of fear memory. From a neurological perspective, PTSD is interesting because the implicated functional neural circuit dysregulation aligns with the known function of the affected brain regions across species, in neuroimaging studies and in translational neuroscience studies.
Engram. A theoretical representation of a neural unit of memory storage. |
The majority of research into the neuroanatomy of PTSD has focused on the role of the amygdala and its subregions in fear and threat processing (FIG. 1b). We now know that sensory information forming the representation of the conditioned stimulus is received in the lateral and basolateral nuclei of the amygdala and integrated with aversive and pain information (the unconditioned stimulus), leading to the consolidation of threat memory via long-term potentiation-like enhancement of synaptic efficacy. Similarly, fear memory consolidation depends upon numerous molecular mediators of plasticity, including glutamatergic NMDA-dependent mechanisms, BDNF, calcium-dependent mechanisms and CREB-dependent changes in gene expression. Together, these events lead to enhanced synaptic activity and long-term structural changes within the amygdala, such that future activations of the conditioned-stimulus sensory engram alone become sufficient to activate many of the downstream pathways that were previously activated only by the unconditioned stimulus.
The results of several decades of research into the downstream pathways of the amygdala — in multiple species, including rodents, non-human primates and humans — indicate that hard-wired axonal projections from neurons within the central–medial subdivision of the amygdala lead to many of the ‘fear’ and ‘panic’ reflexes that are observed during a traumatic cue-induced or trigger-induced panic response (FIG. 1c). These reflexes include increased heart rate mediated by projections to the hypothalamus, locus coeruleus and dorsal vagal nerve, increased respiratory rate via parabrachial connections, gastrointestinal distress via dorsal vagal connections, increased startle via projections to the RPC, freezing and social anxiety via projections to the periaqueductal grey, and hypothalamic–pituitary–adrenal (HPA) activation via projections to the paraventricular nucleus of the hypothalamus. Thus, the fear-induced and threat-induced activation of threat responses are among the most well understood ‘behavioural reflexes’ in neuropsychiatry.
The hippocampus has been implicated in PTSD since the earliest neuroimaging studies of the disorder. Multiple studies, beginning with one by Bremner et al. in 1995, have reported smaller hippocampal volumes in individuals with chronic PTSD than in healthy control participants, and this finding has now been replicated in large-scale neuroimaging meta-analyses. As outlined in more detail below, the roles of the hippocampus in context modulation of fear memory responses and in discrimination versus generalization of threat-related cues and contexts are all thought to be relevant to PTSD formation and maintenance. One of the long-standing questions related to reduced hippocampal volumes and PTSD pertains to the issue of cause versus effect. Notably, multiple preclinical studies found an association of trauma and chronic stress with smaller hippocampal volume. However, pre-existing hippocampal deficits in model systems are associated with an increased risk of stress responses. Thus, less robust hippocampal structure and/or function could be a pre-existing risk factor for the development of PTSD following subsequent trauma. Consistent with this directionality, evidence from human and animal studies indicates that the hippocampus has a clear role in the extinction, or learned inhibition, of cued fear memories, and that hippocampal disruption might be important for the extinction deficits seen in PTSD.
The medial prefrontal cortex, in particular the subgenual prefrontal cortex, in humans is thought to be relatively homologous to the infralimbic region in the rodent brain and is increasingly being implicated in the neurobiology of PTSD. In both rodent studies and human studies of fear inhibition and PTSD, this brain area seems crucial — working in concert with the hippocampus — in providing inhibitory control over threat-related memories and behaviours (FIGS 2,3). Decreased subgenual prefrontal cortex activation and reduced white matter integrity of the uncinate fasciculus, which connects medial prefrontal cortex regions to the amygdala and other anterior subcortical structures, have been observed in individuals with PTSD compared with healthy control participants. By contrast, the dorsal anterior cingulate cortex (dACC) within the medial prefrontal cortex seems to be relatively homologous to the rodent prelimbic cortex and both areas have been implicated in increased fear-responding and threat-responding, and are often co-activated with the amygdala during the threat response.
Importantly, in regions associated with regulation of arousal and emotion, the dissociative subtype of PTSD tends to be associated with opposite patterns of brain activation than the ‘classic’ pattern of PTSD described above. In general, individuals with dissociative PTSD have a pattern of ‘emotional overmodulation’, with increased activity in the rostral anterior cingulate and medial prefrontal cortex, areas of the brain that are generally involved in regulating emotion and arousal. By contrast, individuals with PTSD without substantial dissociation demonstrate ‘emotional undermodulation’ with decreased activity in the aforementioned areas. Importantly, large-scale functional network connectivity seems to be dysregulated in individuals with PTSD and dissociation, such that trauma-related dissociative symptoms, distinct from PTSD and childhood trauma, can be estimated on the basis of network connectivity. These clinical and neurobiological findings provide consistent support for the inclusion of a dissociative subtype of PTSD in diagnostic nomenclature.
In summary, ‘classic’ PTSD is associated with increased threat-responding, hyperarousal, hypervigilance and intrusive trauma-associated memories. Furthermore, cohort studies have repeatedly found increased amygdala, insula and dACC activation to threatening cues as well as decreased hippocampal and subgenual prefrontal cortex activation in individuals with the disorder. These findings are consistent with a model in which cue-related threat-responding is dysregulated and hyperactivated and is not subject to normal inhibitory suppression via safety contexts and extinction memory formation (FIGS 1,2a–d). A somewhat opposite pattern of brain activity has been reported in individuals with the dissociative subtype of PTSD, suggesting fundamentally different pathophysiology.
The neurobiology of PTSD symptoms
Sleep disturbances.
One of the earliest signs of PTSD is sleep disturbance, which often includes nightmares, insomnia and fragmented sleep architecture. As is the case with hippocampal size, sleep difficulties might be both a risk factor and a symptom of PTSD. Studies in military and civilian populations have reported an association between the presence of sleep problems prior to trauma and increased PTSD risk following trauma. Notably, sleep disturbances sometimes persist after other PTSD symptoms subside with treatment. The sleep symptoms of PTSD vary across individuals but many people with PTSD have trouble falling asleep and wake easily, often waking up many times at night. Intrusive memories, in the form of nightmares, are a classic symptom of PTSD, and serve to both exacerbate overall PTSD symptoms and contribute to disrupted, non-refreshing sleep. The content of these nightmares often relates to details of past trauma, with many individuals with PTSD reporting repetitive nightmares. Post-traumatic nightmares can be treated with imagery rehearsal therapy, which involves the patient ‘rewriting’ the script of the dream with a less threatening version during a therapy session. This type of therapy is thought to provide cognitive reframing together with a form of exposure-based extinction recovery from the negative traumatic memories experienced through the nightmares, which is similar to the approach used with other trauma-informed therapies.
Rates of extinction and safety learning seem to partially explain the difference between people who are resilient and able to recover from a traumatic event compared to those who maintain acute stress responses and develop PTSD. As discussed above, compared with healthy individuals, people with PTSD have been found to have higher ‘fear load’ during extinction, worse extinction learning, poorer extinction recall and worse safety learning. Notably, some data from studies in humans suggest that extinction deficits are mediated in part by fragmented rapid eye movement (REM) sleep. Therefore, future studies could benefit the field by examining relationships between emotional learning and disturbed sleep in PTSD. This finding also raises the possibility that sleep status surrounding the traumatic exposure could be a factor in pathogenesis and hence a target for mitigation or prevention.
Orbicularis muscle. A muscle located in the eyelid, activity of which is often an end point in human fear conditioning research. |
|---|
Endophenotypes. Secondary phenotypes that reliably co-occur as a sub-feature of a broader primary phenotype. |
With regards to the neural circuitry of PTSD, the hippocampus, amygdala, dACC and insular cortex are all implicated in sleep disturbance (FIGS 1–3). As discussed above, these brain regions are thought to be responsible for causing the individual with PTSD to revisit the traumatic event in flashbacks and nightmares and for maintaining a state of hyperarousal. When compared with healthy control participants, individuals with PTSD had a faster heart rate while sleeping, indicating the presence of an enhanced threat response that keeps the body in an overall state of hypervigilance. Notably, the hallmarks of disturbed sleep in PTSD include more time spent in stage-one light sleep, less restorative slow-wave sleep and fragmented REM sleep. Some of these core features have also been observed in rodents exposed to traumatic stress. Disruptions in the above brain circuits, combined with dysregulated activity of brainstem activating systems (for example, locus coeruleus and periaqueductal grey) are thought to contribute to abnormal sleep patterns and increased nightmares in PTSD. Studies in animal models have demonstrated that stress-induced changes in the function of specific cell populations within the nucleus accumbens, a brain area classically implicated in motivated behaviour and regulation of mood, can produce alterations in sleep architecture, providing a putative neural basis for comorbidity in key features of stress-related illness.
Hypervigilance and hyperarousal.
Individuals with PTSD seem to have hypervigilance associated with the acute-threat behavioural system. Acute threat, which encompasses the concept of fear, is defined as activation of the brain’s defensive motivational system to promote behaviours that protect the organism from perceived danger (BOX 1). Fear or threat responses are among the most common and consistent underlying factors of PTSD and a number of other trauma-related disorders. For example, individuals with PTSD often describe that they almost never feel ‘safe’. Instead, they feel acutely threatened by unexpected and generalized cues, and this sense of fear and threat pervades much of their lives, leading to the avoidance of potential contexts and cues that could activate the threat response system. Prolonged activation of the threat response — sustained threat — in PTSD is thought to occur in part via ongoing inescapable intrusive thoughts, flashbacks and nightmares. Furthermore, the active avoidance of cues, contexts and other reminders associated with the trauma means that individuals with PTSD are unable to naturally extinguish the initial fear responses. Numerous factors, such as enhanced amygdala activity and decreased ‘top-down’ cortical regulation, have been associated with fear and threat dysregulation, increased trauma load, and decreased recovery from fear.
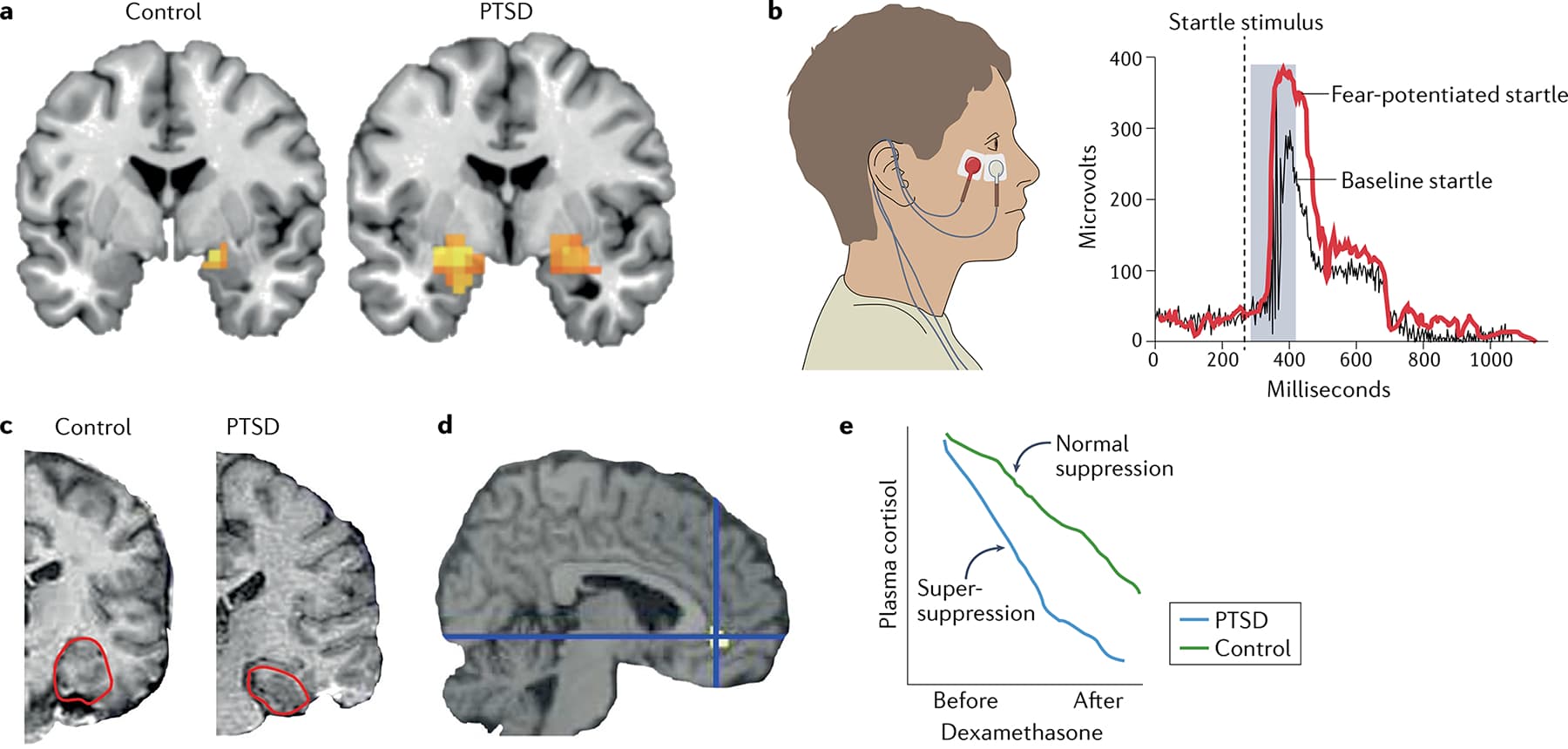
One way of assessing vigilance is by studying the acoustic startle response. For example, while at home and in a state of calmness, healthy individuals might exhibit a slight twitch in response to a loud unexpected noise. However, if the same decibel level of unexpected noise was encountered in a dark alley or at another time of increased vigilance, the startle response would be much amplified. Many individuals with PTSD are always in such a state of hypervigilance and exhibit an increased startle response, which is often described by these individuals as a state of being ‘jumpy’ or ‘overly reactive’ to any slight or unexpected noise. In laboratory settings, this response can be studied in humans by measuring the eyeblink startle reflex (FIG. 2d). This reflex is assessed by measuring the electrical activity of the orbicularis muscle during the presentation of different unexpected auditory cues in the presence of threatening or safe conditions. Numerous laboratory studies have found that individuals with PTSD have enhanced anticipatory startle responses and enhanced fear cue-related startle responses compared with healthy participants and participants who experienced trauma but did not develop PTSD.
The neural circuitry underlying the acoustic startle reflex is well understood — direct projections from the auditory brainstem and thalamic nuclei to the reticularis pontis caudalis (RPC) activate spinal motor pathways, thereby eliciting a rapid muscle extension–flexion response. This circuitry was characterized over several decades by Davis and colleagues, who found (in rats and in humans) that central amygdala projections to the RPC ‘gate’ the startle response to an auditory cue. They also demonstrated that, in a high threat-responsive state, increased activation of amygdala–RPC projections contributes to elevated startle responses.
Additionally, evidence from functional MRI studies indicates that PTSD comprises endophenotypes (also known as intermediate phenotypes) such as enhanced amygdala activation to fearful cues, impaired ‘top-down’ inhibition between the prefrontal cortex and the amygdala, and reduced rostral anterior cingulated cortex activation during emotional processing. These data suggest that hyperactivation of threat salience networks, in particular the amygdala, dACC and insula, in the early aftermath of trauma and during the early recovery period as well as with chronic PTSD are all associated with ongoing hypervigilance and increased threat responses.
Arousal refers to the sensitivity of the organism to external and internal stimuli and exists along a continuum. Arousal facilitates interaction with the environment in a context-specific manner, can be evoked by external (environmental) or internal stimuli, and represents an activated physiological state that is often accompanied by corresponding elevations in threat assessment (hypervigilance). The degree of arousal is indicated by the degree of sympathetic nervous system activity, which is frequently measured using heart rate, skin conductance and the aforementioned eyeblink startle reflex. Increased heart rate and skin conductance in response to trauma imagery, indicative of increased arousal, have been consistently demonstrated in individuals with PTSD compared with healthy control participants. In addition, elevated physiological responses, such as increases in the acoustic startle reflex, have been observed in individuals with PTSD and can serve as a biomarker of the development of sustained heightened arousal. These observations support the theory that the development of sustained heightened arousal in PTSD is characterized by progressive neuronal sensitization, and that dysregulation in sympathetic nervous system arousal, particularly heart rate, skin conductance and eyeblink in response to startling stimuli, might be an endophenotype of the disorder. Notably, data from large prospective studies suggest that the presence of such sensitization in patients in the emergency room predicts the subsequent development of PTSD. These data indicate that elevated skin conductance and eyeblink startle are markers of dysregulated arousal that predates the trauma exposure and/or is a phasic response to acute trauma.
Co-regulated. Two or more biological processes that are modulated (activated, suppressed) in parallel by a common upstream factor. |
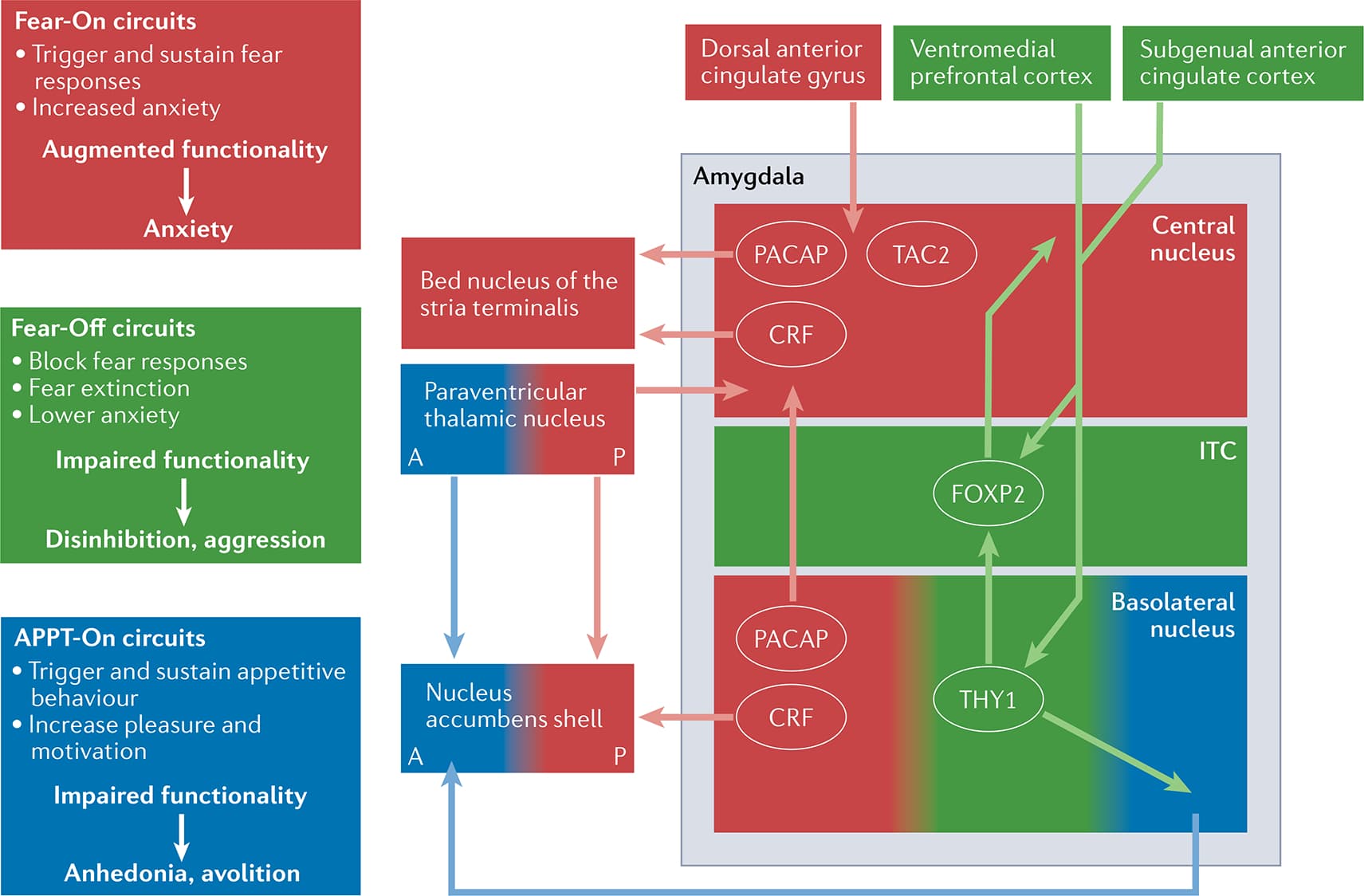
Although an exhaustive discussion of the neuroendocrinology of PTSD is beyond the scope of this Review, repeated studies have demonstrated abnormal regulation of the HPA stress axis (which regulates endocrine function and emotional responses) in PTSD. As an example, data on baseline levels of adrenocorticotrophic hormone (commonly referred to as ACTH) and cortisol in individuals with PTSD are somewhat variable, but multiple studies have identified a PTSD-associated hypersensitivity to HPA feedback at the level of the pituitary and adrenal gland. That is, dexamethasone suppression tests often show a ‘super-suppression’ of plasma cortisol in participants with PTSD compared with healthy participants and participants with depression (FIG. 2e). This hypersensitivity of the peripheral stress axis is thought to be related to chronic hyperactivity of the CNS upstream signals, for example, corticotropin-releasing factor (CRF), in the amygdala, bed nucleus of the stria terminalis and hypothalamic paraventricular nucleus (FIG. 3). Although CRF antagonists have not been not successful in treating PTSD in clinical trials, the underlying biology and clinical presentation of PTSD is clearly variable, and behavioural, physiological and/or blood-based biomarkers for stratifying specific biological subtypes of PTSD will be crucial for success with targeted therapeutics.
Cognition and memory deficits.
Although deficits in numerous aspects of cognition and memory are seen in PTSD, declarative memory is particularly impaired when the index trauma is accompanied by comorbid traumatic brain injury (TBI). TBI is often but not invariably present in individuals with PTSD. One hypothesis is that brain injury-related processes (inflammation, cell death) exacerbate the molecular adaptations that occur in response to non-injury-related stress. Deficits in declarative memory also frequently accompany an increased vulnerability to PTSD in individuals who have experienced a natural disaster or motor vehicle accident. The brain region most associated with PTSD-related declarative memory deficits is the hippocampus, which is involved in memory formation, storage and consolidation. Notably, some of the oldest data on hippocampal structure indicate smaller hippocampal volumes in individuals with PTSD than in control participants. These findings have now been replicated in a much larger meta-analytic study. In other studies, smaller hippocampal volume at 1-month post-trauma and decreased inhibition-related hippocampal activity both predicted PTSD severity at later time points. These data provide evidence that hippocampal volume before PTSD development is inversely correlated with the likelihood of later development of PTSD.
Insights from omics studies
Post-mortem brain tissue.
Numerous research teams are currently examining molecular findings in PTSD in post-mortem human brains. The largest analysis to date was published in 2021 by Girgenti and colleagues, who performed differential gene expression and network analyses on transcriptomic data from four prefrontal cortex regions from participants with PTSD. They found that a co-regulated set of genes marking interneuron function was downregulated in the brains of individuals with PTSD compared with those of healthy control participants, representing the most significant gene network alteration associated with PTSD. They then integrated these transcriptomic data with large-scale genome-wide association study (GWAS) data, identifying an association between expression of the interneuron synaptic gene ELFN1 and genetic liability for PTSD. Additional analyses found that differential sexually dimorphic transcriptomic regulation might contribute to the higher rates of PTSD in women. This analysis provides an initial level of convergence between prefrontal cortex gene expression pathways and large-scale genetic findings, suggesting that dysregulation of inhibitory cortical circuits is critical to the pathophysiology of PTSD in humans.
Another study identified an association between multiple forms of psychopathology and advanced DNA methylation age. The results of several studies have suggested that PTSD and other stress-related disorders increase the risk of neurodegenerative diseases. Using PET imaging, Mohamed and colleagues found that, compared with healthy control participants, participants with PTSD with and without a history of TBI had widespread tau accumulation in neocortical regions that overlapped with typical and atypical patterns of Alzheimer disease-like tau distribution. They also found evidence for advanced epigenetic ageing in the brain tissue of individuals with PTSD. Before the introduction of current multi-omic approaches, several studies had identified changes in the expression of plasticity-related genes in individuals with PTSD. In particular, Licznerski and colleagues examined post-mortem samples of dorsolateral prefrontal cortex from individuals who had undergone traumatic stress. They found that expression of the gene encoding serum and glucocorticoid regulated kinase 1 (SGK1) was downregulated in participants with PTSD compared with participants without PTSD. They validated this finding preclinically by showing that inhibition of SGK1 in the medial prefrontal cortex of rats results in helplessness-like and anhedonic-like behaviours and abnormal dendritic spine morphology and synaptic dysfunction. A number of additional, larger post-mortem studies are in progress, and the results of these will rapidly expand our understanding of the transcriptomic, epigenetic and proteomic landscape of the human brain in PTSD.
Peripheral biomarkers.
In addition to work on post-mortem brain samples, biomarker identification from the peripheral tissue has also proven feasible in PTSD research, leading to many new discoveries. Examples include the large-scale genetics studies and GWAS, outlined in more detail below, that have begun to identify the genetic architecture of PTSD. Furthermore, hormonal measures, such as the reproducible findings of super-suppression of the cortisol–HPA axis mediated by FKBP5 and findings of enhanced inflammation in PTSD have all been robust and important findings for understanding PTSD biology. New integrative studies of multi-omics in the aftermath of trauma are also providing powerful predictive biomarker approaches. Finally, peripheral epigenetics, in the form of studies of epigenetic ageing and identification of novel cell signalling pathways, as well as the demonstration of shared epigenetic markers across blood and brain, are pointing towards new leads in understanding PTSD.
GWAS.
Identifying genetic alterations in the biological pathways that mediate arousal and stress might reveal variations that make some individuals more vulnerable than others to the effects of stress or trauma exposure and, hence, to the development of PTSD. The past decade has witnessed a rapid expansion in our understanding of the genetics of PTSD, with large-scale consortia, including the Psychiatric Genomics Consortium (PGC), UK Biobank and the US Million Veterans Program (MVP), performing GWAS of tens of thousands of individuals with PTSD and hundreds of thousands of controls. These efforts have combined with a revitalization of post-mortem studies, using modern transcriptomics and proteomics, as well as new single-cell RNA sequencing approaches. As a result, the field is starting to see the convergence of some PTSD-associated molecular pathways and genetic alterations on the neural circuit regions that underlie the threat response.
Several large-scale GWAS studies of PTSD have been performed to date. As these ongoing studies continue and the sample sizes increase at each intermediate (‘freeze’) analysis, several robust genome-wide significant loci have been associated with PTSD. The PGC-PTSD working group anticipates that many more genome-wide significant loci will have been identified by early 2022 in a planned analysis (termed ‘freeze 3’) of hundreds of thousands of samples. Notably, many of the significant PTSD-associated genes identified thus far, including those involved in sensitivity to the stress peptide CRF (see below), are expressed in brain circuits previously implicated in PTSD. Furthermore, preliminary data from post-mortem brain studies of participants in the PGC-PTSD GWAS cohort suggest that some of the gene pathways will overlap with differentially expressed genes that have been identified in other PTSD post-mortem studies.
Two of the largest published GWAS to date come from the MVP. Stein et al. conducted genome-wide association analyses of over 250,000 MVP participants using electronic health record-validated data on PTSD diagnosis and quantitative symptoms. Three significant loci were identified in case–control analyses of participants of European ancestry and 15 significant loci were identified in quantitative symptom analyses. The combination of these findings with heritability analysis suggested enrichment in several cortical and subcortical regions. Previous analyses of the same cohort by Gelernter et al., published in 2019, examined genetic data from ~147,000 American individuals of European ancestry and ~20,000 African American individuals in the MVP to identify risk factors relevant to intrusive re-experiencing of trauma — the most characteristic symptom cluster of PTSD. In American individuals of European ancestry, eight distinct significant regions were identified, of which three (CAMKV, TCF4 and a chromosome 17 locus including KANSL1 and CRFR1) were highly significant (P < 5 × 10−10). The association between intrusive re-experiencing of trauma and CRFR1 is particularly relevant given the previous findings that indicate a role for a dysregulated HPA axis in PTSD and interest in CRF antagonists as therapies for certain subtypes of PTSD. Overall, the results from these well-powered GWAS provide new insights into the biology of PTSD.
The PGC-PTSD working group also performed a GWAS in a multi-ethnic cohort. This analysis included data from more than 30,000 participants with PTSD and 170,000 control participants. The results confirmed previous PTSD heritability estimates of 5–20%, varying by sex. The genes highly significantly associated with PTSD included novel genes and non-coding RNAs as well as PARK2, which has been previously implicated in Parkinson disease and is involved in dopamine regulation. Using a partially overlapping data set from the PGC-PTSD GWAS, Huckins et al. used brain and non-brain transcriptomic imputation to identify genetically regulated gene expression in ~30,000 participants with PTSD and ~166,000 control participants. They found 18 significant genetically regulated gene expression–PTSD associations corresponding to specific tissue–gene pairs. Of particular interest, Huckins et al. found that the expression of SNRNP35, a gene critical for RNA splice regulation, is dependent on both corticosteroids and stress, and is predicted to be downregulated in the dorsolateral prefrontal cortex of individuals with PTSD. Together, these results further demonstrate a role for genetic variation in the biology of PTSD risk.
In early 2022, the MVP and PGC-PTSD data will be merged for a meta-analysis, termed freeze 3, and on the basis of the increased power provided by hundreds of thousands of additional samples, many more genome-wide significant loci are expected. Thus, ongoing genetics analyses, combined with functional transcriptomics and proteomics, are leading to the identification of important new insights into the genetic basis of PTSD that can be integrated with our neural circuit-based understanding of trauma-related dysfunction that characterizes this condition.
Key pathways.
Understanding the ways in which the risk genes described above contribute to the development and persistence of PTSD requires parallel studies of the brain pathways regulated by these genes. Two key stress pathways that have emerged as particularly relevant candidate moderators of risk, clinical presentation and neurobiological characteristics of PTSD are the CRF and pituitary adenylate cyclase-activating polypeptide (PACAP) systems. Similar to evidence noted above indicating that levels of CRF and peptides involved in the HPA axis are altered in individuals with PTSD, evidence exists of higher circulating blood levels of PACAP in individuals with PTSD, especially women, than in individuals without PTSD. Moreover, allelic variation in the genes encoding the type 1 receptors of CRF and PACAP (that is, CRFR1 and PAC1R) predicts the presence of greater hyperarousal symptoms and total symptoms of PTSD as well as greater physiological arousal during stress-related and anxiety-related paradigms. Importantly, CRFR1 and PAC1R are richly expressed within the components of canonical threat brain circuit in PTSD, including in the amygdala, bed nucleus of the stria terminalis and medial prefrontal cortex. Taken together, these data suggest that the CRF and PACAP systems contribute to the differential risk of PTSD in women versus men and to neural alterations that mediate fear and hyperarousal in PTSD. Understanding the similarities and differences between the acute and persistent effects of these peptides may offer new methods of diagnosing and treating PTSD.
One of the most studied molecular mechanisms underlying stress-related pathophysiology is the FKBP5 pathway, which regulates the glucocorticoid response within cells. Variation in the FKBP5 gene was first identified in individuals with PTSD who experienced abuse as children and, since then, changes in FKBP5 expression have been linked to many aspects of PTSD pathophysiology, including the type and severity of symptoms, neural activity, and startle physiology. Studies in animal models of PTSD have also consistently pointed to a role for FKBP5 in traumatic stress. Additionally, post-mortem studies have now identified increases in FKBP5 expression in multiple cortical regions in individuals with PTSD compared with control participants. Although FKBP5 has yet to be identified on a large-scale GWAS of PTSD, these compelling findings suggest that it is likely to be important in gene–environment regulation of the stress response.
Therapeutics: treatment and prevention
The current standard treatments for PTSD include pharmacotherapies and psychotherapies. In general, pharmacotherapies reduce symptoms related to anxiety, arousal and depression. Evidence-based psychotherapy approaches range from supportive and emotional skills-building to exposure-based therapies that aim to restructure the underlying dysregulated traumatic memories. Currently, the only FDA-approved treatments for PTSD are the serotonin reuptake inhibitors sertraline and paroxetine; however, numerous serotonergic, dopaminergic and noradrenergic antidepressants and/or anxiolytic medications have shown some efficacy for relieving the symptoms of PTSD in double-blind, randomized placebo-controlled trials (RCTs).
Although dopamine receptor D2 antagonists (for example, atypical antipsychotic drugs) have some utility for the treatment of refractory PTSD that is non-responsive to other treatments, including first-line serotonin reuptake inhibitor treatment, the largest RCTs of the D2 risperidone augmentation failed to show a benefit of treatment. Specifically, open-label trials and small RCTs have reported that treating patients with trauma-related intrusive thoughts and sounds/voices with atypical antipsychotic medications can be particularly helpful. The results of two meta-analyses suggest that low-dose atypical antipsychotic medications can be useful for augmentation in refractory PTSD comorbid with depression, similar to the beneficial effects of these drugs in refractory depression. In small trials, anti-epileptic drugs that are used as mood stabilizers (for example, sodium valproate, topiramate and lamotrigine) have also been found to have some efficacy in PTSD, in particular related to mood and anger dysregulation; however, larger-scale RCTs of these drugs did not show robust effects.
The medication that could be considered to have the most ‘precision’ target in PTSD is prazosin, an α-adrenergic antagonist, that has been shown in several RCTs to decrease the occurrence of nightmares in PTSD. Prazosin was originally administered to veterans with PTSD for the treatment of hypertension or benign prostate hypertrophy, which is a frequent comorbidity of PTSD, and was found to also help with nightmares. The drug was then repurposed for use in PTSD on the basis of its targeting of subcortical α1-adrenergic receptors that are involved in emotional hyperarousal and norepinephrine-mediated sleep dysregulation. Early small trials of prazosin in individuals with PTSD produced very promising results, and moderately powered randomized trials have reported benefits of prazosin over placebo on trauma nightmares, sleep quality and total PTSD symptoms. Unfortunately, the largest RCT to date, published in 2018, failed to identify an effect of prazosin on nightmares or sleep quality in veterans with PTSD. However, prazosin is known to have a narrow therapeutic window in terms of dose, and doses similar to those required for an effect on nightmares have an effect on orthostasis, which makes the use of higher doses untenable. Therefore, an optimal dose for an effect on nightmares might have not been reached in this study. Furthermore, no biomarkers of adrenergic dysregulation, which might identify individuals who would be most responsive to this treatment, were required for study inclusion. Prazosin treatment could still be a useful approach but, as with many treatments for PTSD and other psychiatric indications, identifying biomarkers of treatment efficacy for patient stratification will be critical given the vast heterogeneity of the syndrome.
To date, the most efficacious treatment for PTSD has been trauma-focused psychotherapy, generally in the form of exposure-based treatments. With ‘imaginal exposure’, a patient describes the experience of the traumatic event in as much detail as possible to the therapist. The patient then repeatedly re-tells this memory over extended periods of time; indeed, the most common exposure-based treatment regimen is referred to as prolonged exposure. Through this process, over multiple therapy sessions, with each one focused on the most salient distressing memory at the time, the patients’ emotional distress to the memory diminishes. Patients often describe feeling as if a ‘black hole’ of negative memory and emotion becomes neutralized, if not almost boring to them. This process of exposure is thought to diminish fear via the well-understood neural mechanisms of Pavlovian conditioning-based ‘extinction’ learning described above. Extinction can be conceptualized as ‘retraining’ the brain, specifically through known neural circuits that mediate threat responses so that previously highly threatening cues are now re-learned — and experienced — as signalling safety. On the basis of animal studies described above, it seems likely that extinction plays a key role in successful prolonged exposure therapy and other similar cognitive behaviour therapies such as cognitive processing therapy and eye movement-desensitization and reprocessing therapy, which are also common trauma-focused psychotherapies for PTSD.
Possible future approaches.
Considering the rapid progress in our understanding of the neurobiology and biomarkers that might predict trajectories of PTSD, we expect that future approaches to treatment will leverage these neurobiological and biomarker targets. Approaches in development include the combination of pharmacological targeting of neural plasticity and targeted emotional learning as well as EEG-based biofeedback targeting amygdala activation, both of which aim to specifically enhance the natural learning processes that underlie fear inhibition and extinction. Novel pharmacological therapies targeting the cellular and molecular pathways identified in genetic, transcriptomic and translational studies are also being developed. Additionally, other experimental treatments, including ketamine derivatives and drugs that block kappa-opioid receptors, show evidence of being able to mitigate stress responses in animal models of PTSD if given prophylactically, raising the intriguing possibility that it might someday be possible to prevent the development of PTSD. Although stress can be unpredictable in the context of everyday life, some of the most severe, debilitating and costly forms of stress (for example, those encountered during a combat mission or while responding to a disaster) involve a recognizable ‘lead time’ that precedes exposure, offering a window of opportunity for prevention.
Conclusions and future directions
This Review addresses the neuroscience-based understanding of some of the primary symptoms of PTSD, including hyperarousal, dissociation, intrusions and sleep dysregulation, all of which are increasingly being understood through translational research. Evidence suggests that PTSD can be viewed as a disorder that involves dysregulation of normal fear processes, and the neural circuitry underlying fear and threat-related behaviour and learning in mammals has been defined in great detail over the past 40 years. The underlying circuitry includes hub brain regions such as the amygdala, insula, hippocampus and the medial prefrontal circuit, and these are among the most well-understood brain circuits in behavioural neuroscience. Notably, the study of threat responses and their underlying circuitry has led to rapid progress in our understanding of learning and memory processes. Finally, large-scale genetic approaches to understanding trauma-related disorders and PTSD have been highly successful. These findings, along with those from transcriptomics, metabolomics and proteomics studies, are rapidly expanding the list of potential targets for personalized medicine and patient stratification. The next few years offer great promise for combining genetic discoveries with a deep understanding of the neural circuits that regulate the core behavioural features of PTSD.
In conclusion, PTSD is a syndrome that is common in individuals who have been exposed to severe trauma, is frequently comorbid, and is associated with a significantly increased risk for morbidity and mortality. The integration of advances in our understanding of the neural circuitry, physiology, intermediate phenotypes and genetics of PTSD, along with large-scale longitudinal studies, offer great promise for progress in the prediction, intervention and, possibly, prevention of this debilitating psychiatric disorder.
Key points.
Post-traumatic stress disorder (PTSD) is a debilitating neuropsychiatric disorder, characterized by re-experiencing, avoidance, negative emotions and thoughts, and hyperarousal.
PTSD is frequently comorbid with neurological conditions such as traumatic brain injury, post-traumatic epilepsy and chronic headaches.
PTSD has a prevalence of approximately 6–8% in the general population and up to 25% among individuals who have experienced severe trauma.
Many of the neural circuit mechanisms that underlie the PTSD symptoms of fear-related and threat-related behaviour, hyperarousal and sleep dysregulation are becoming increasingly clear.
Key brain regions involved in PTSD include the amygdala–hippocampus–prefrontal cortex circuit, which is among the most well-understood networks in behavioural neuroscience.
Combining molecular–genetic approaches with a mechanistic knowledge of fear circuitry will enable transformational advances in the conceptual framework, diagnosis and treatment of PTSD.