Abstract
Opioids are a mainstay in acute pain management and produce their effects and side effects (e.g., tolerance, opioid-use disorder and immune suppression) by interaction with opioid receptors. I will discuss opioid pharmacology in some controversial areas of enquiry of anaesthetic relevance. The main opioid target is the µ (mu,MOP) receptor but other members of the opioid receptor family, δ (delta; DOP) and κ (kappa; KOP) opioid receptors also produce analgesic actions. These are naloxone-sensitive. There is important clinical development relating to the Nociceptin/Orphanin FQ (NOP) receptor, an opioid receptor that is not naloxone-sensitive. Better understanding of the drivers for opioid effects and side effects may facilitate separation of side effects and production of safer drugs. Opioids bind to the receptor orthosteric site to produce their effects and can engage monomer or homo-, heterodimer receptors. Some ligands can drive one intracellular pathway over another. This is the basis of biased agonism (or functional selectivity). Opioid actions at the orthosteric site can be modulated allosterically and positive allosteric modulators that enhance opioid action are in development. As well as targeting ligand-receptor interaction and transduction, modulating receptor expression and hence function is also tractable. There is evidence for epigenetic associations with different types of pain and also substance misuse. As long as the opioid narrative is defined by the 'opioid crisis' the drive to remove them could gather pace. This will deny use where they are effective, and access to morphine for pain relief in low income countries.
Opioids are a mainstay for pain management in the perioperative period. That they are effective in a range of types of nociceptive pain is clear where they modulate nociceptive information flow. Their use/efficacy more widely in chronic pain is controversial, particularly in neuropathic pain, but there is utility in the palliative care setting. Alongside the beneficial analgesic actions (antinociception in animals) opioids produce a troublesome set of adverse effects; these include ventilatory depression, constipation, immune suppression, tolerance and opioid use disorder. Opioid-induced hyperalgesia is also a significant clinical problem. Tolerance is often viewed at the centre of an adverse effect circle where increased dosing is required, but this produces more tolerance (and the other adverse effects). The focus of this review is to explore improved analgesia, but it is important to remember that tolerance can manifest secondary to disease progression (pseudo-tolerance) and can develop to many adverse effects; this includes ventilatory depression.
Opiates are natural products from the poppy but also encompass natural endogenous peptides (endorphins for example) whereas opioids are synthetic and not found in nature. Moreover, it is important to remember that not all opioids used in the clinic are the same. At therapeutic concentrations the vast majority display μ (mu, MOP) receptor selectivity. However, there is an approved synthetic peptide κ (kappa, KOP) receptor agonist difelikefalin, but this is used as a treatment for pruritis associated with chronic kidney disease. There are marked differences in clinical MOP ligands with respect to receptor potency (e.g. fentanyl is ∼80× more potent than morphine), efficacy (e.g. buprenorphine is a partial agonist) and in pharmacokinetics (e.g. rapid metabolism for remifentanil). If we consider immune suppression, morphine is strong, oxycodone is weaker and buprenorphine has almost no activity; clearly with respect to immune suppression opioids are not all the same. As covered below in in vitro studies, opioids at ‘equipotent’ concentrations can activate different signalling pathways. Considering the above arguments regarding marked differences in opioid pharmacological behaviour, Emery and Eitan state “… different opioids cannot be made equivalent by merely dose adjustment” other approaches are required.
In this focused review I will cover opioid receptors and mechanisms in some controversial areas of enquiry in a digestible format and use this to frame attempts to design safer opioid medications.
Opioid receptors
Opioid receptors are class A G protein-coupled receptors (GPCRs) and are part of a family; these are the classical naloxone-sensitive MOP, δ (delta, DOP) and KOP along with the non-classical receptor for nociceptin/orphanin FQ (N/OFQ) or NOP (Fig 1). The consequences of receptor interactions will be discussed later. There is historic pharmacological evidence to suggest subtypes of opioid receptors and we have reviewed this in the past. The observation that knock-out of a single gene for each receptor results in full loss of function argues against subtypes but there is evidence for splice variants; with extensive data for MOP. These splice variants can explain some differences in function but not the pre-cloning pharmacological suggestion of subtypes. Moreover, there are now knock-in animals expressing trackable genetic variants to address basic pharmacological-neurobehavioural responses. These animals have natural opioid gene sequence replaced with a modified sequence typically also encoding a fluorescent probe so that expression can be tracked or containing a single nucleotide polymorphism.
Figure 1.
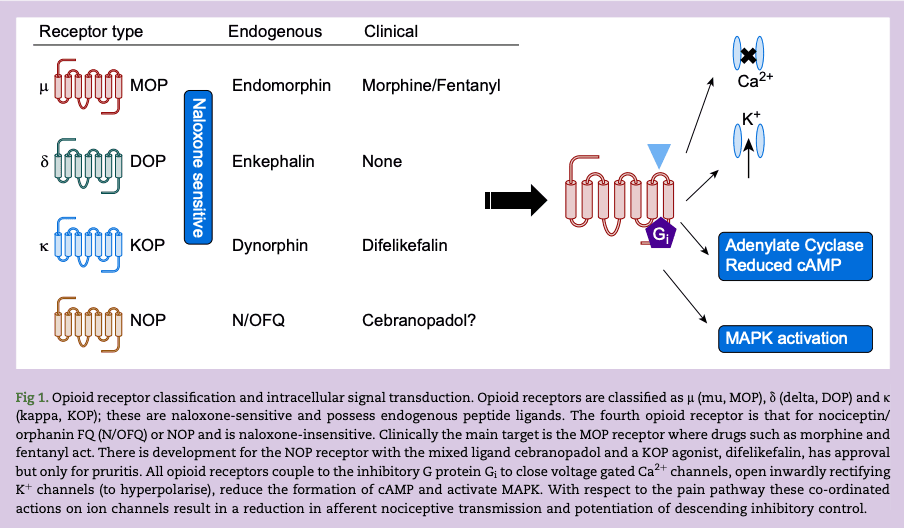
All opioid receptors couple via the inhibitory heterotrimeric G protein (composed of α and β/γ subunits), Gi. Binding of the opioid ligand to the orthosteric site, facilitates G protein interaction and guanine nucleotide (guanosine diphosphate [GDP] for guanosine triphosphate [GTP]) exchange on the α subunit which dissociates from the β/γ dimer. The αi-GTP and variably β/γ dimer go on to inhibit adenylate cyclase to reduce cyclic adenosine monophosphate (cAMP), open inwardly rectifying K+ channels to hyperpolarise, close voltage gated Ca2+ channels and activate mitogen-activated protein kinases (MAPKs) (Fig 1). The opioid signal is terminated by GTP metabolism back to GDP (the α subunit is also a GTPase enzyme) and after G protein-coupled receptor kinase (GRK) phosphorylation of the receptor, arrestin recruitment and eventual endocytosis. Arrestin recruitment is important to consider further as there is (disputed) evidence that biased signalling towards G protein and away from arrestin has the potential to produce good quality analgesics with reduced adverse effect profiles.
Opioids modulate both the afferent and efferent parts of the pain pathway. By reducing neurotransmitter release they inhibit pain transmission from first order primary afferent to the second order ascending neurones. These actions are predominantly at K+ and Ca2+ channels where activation of the former enhances K+ efflux leading to hyperpolarisation, while inhibition of the latter reduces Ca2+ influx; both resulting in reduced transmitter release. They also affect second to third order transmission and enhance descending inhibitory control activity; the latter being through reduction in GABAergic inhibitory transmission. With respect to the NOP receptor and pain processing there is significant plasticity.
In a seminal series of papers from the laboratory of Laura Bohn, the involvement of β-arrestin-2 in opioid antinociception and adverse effects was explored. To accomplish this, animals deficient in the gene for β-arrestin-2 production (knock-out or KO animals) were generated. KO animals showed greater antinociception and reduced tolerance. In a further series of studies, KO animals showed reduced ventilatory depression and inhibition of gastrointestinal (GI) motility. The overall proposal was that G protein action was beneficial and β-arrestin-2 action was not; this is the basis of functional selectivity or biased agonism covered below. Ligand bias is when a particular ligand can drive one transduction pathway over another. For the MOP receptor this has been questioned in a study by Kliewer and colleagues where fentanyl and morphine did not display reduced adverse effect profiles in KO animals along with some biased opioid receptor knock-in studies. β-Arrestin-2 bias has also been questioned by Gillis and colleagues with a more simple explanation based on efficacy; putative MOP biased agonists being partial agonists. In a reanalysis of the data from the Gillis paper, Stahl and Bohn conclude ‘The data in the Science Signaling paper provide strong corroborating evidence that G protein signaling bias may be a means of improving opioid analgesia while avoiding certain undesirable side effects’. Whilst of great interest pharmacologically and as a potential driver for early phase drug discovery, we have argued that from a therapeutic (drug to market) perspective it does not matter provided any new ligands provide therapeutic efficacy with low adverse effect profiles.
Opioid pharmaco-therapeutic strategies
Opioid receptors should be viewed as dynamic and existing in multiple conformations (e.g. active and inactive). Rather than being thought of as traditional keys to receptor locks, ligands (opioids) stabilise a particular conformation or the equilibrium between different conformations. Full agonists shift the equilibrium and stabilise the active form whereas partial agonists less so. Neutral antagonists simply block but do not activate receptors. For a receptor to exist in the active form in the absence of agonists it must possess constitutive activity. This can occur via a number of mechanisms and of interest here is a series of inherited diseases associated with variants in genes encoding GPCRs. Ligands that interact with this conformation and reduce activity are inverse agonists; they produce the opposite effect to the standard agonist. In experimental systems there are examples of inverse agonists at MOP, DOP, KOP and NOP; less so for NOP receptors, more so for DOP receptors.
Opioids bind to opioid receptors at the orthosteric ligand binding site and some details of this ligand binding site can be obtained from the crystal structures; details of all four opioid receptors have been resolved. Ligand interaction can modulate receptor function by targeting monomer receptors (the traditional view), dimer receptors (homo and hetero), using allosteric modulators and potentially driving one transduction pathway over another (Fig 2).
Figure 2.
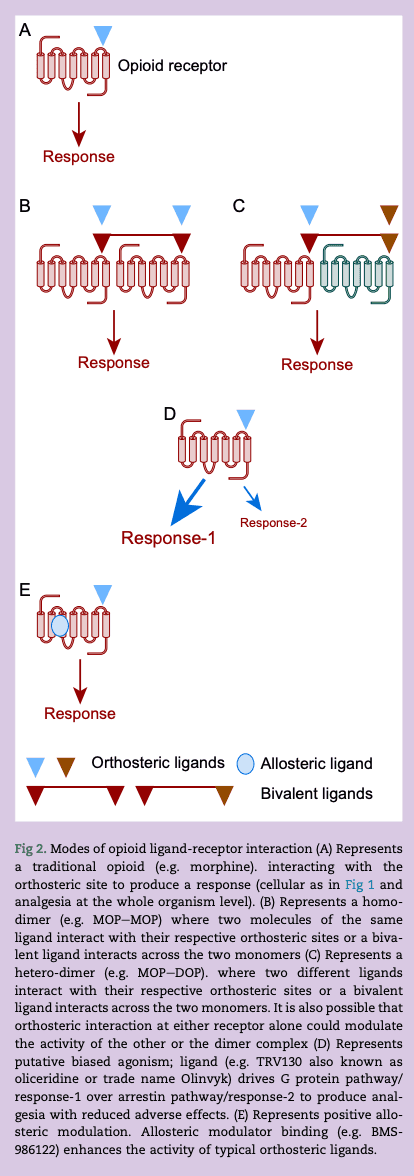
To explore pharmacological classification a little more, in a whole organism context, efficacy is the size or strength of a given response in a particular tissue; for opioids this could be analgesia or ventilatory depression. Agonists that return a lower maximum response than a full (typically endogenous) agonist are partial agonists and have reduced efficacy. Can reduced efficacy be used to therapeutic effect (e.g. buprenorphine) and can differences in efficacy be used to explain the pharmacology of some of the newly produced opioid ligands?
Multi-target strategy
Opioid receptors are unlikely to function alone. There is marked interaction between subtypes and this can be as a result of signalling interaction or the formation of dimers. The existence of opioid homodimers and heterodimers has been known for many years and there are some elegant studies demonstrating this using fluorescent tagged receptors and others using receptor probes. For example, activating MOP whilst inhibiting DOP produces antinociception with a reduced adverse effect profile. This can be accomplished with a MOP agonist (morphine) and a DOP antagonist (naltrindole) or by using a bivalent ligand that interacts with both MOP and DOP simultaneously such as MOP agonist/DOP antagonist UFP-505. Whilst offering much there are no clinically available MOP–DOP bivalents. Moreover, there are data that show that the trivalent opioid agonist DPI-125 with marginal selectivity for DOP over MOP/KOP produces antinociception but with reduced ventilatory depression and abuse liability.
There are other combinations that show potential and MOP–NOP is an example with substantial preclinical and clinical development. Cebranopadol is a mixed NOP-opioid ligand and we have reviewed this molecule in the past. The ligand is a high efficacy partial agonist at NOP and other opioid receptors; MOP being of particular interest here. This mixed (non-selective) opioid is antinociceptive in animal models of nociceptive (tail withdrawal), inflammatory (complete Freund's adjuvant) and neuropathic (nerve constriction) pain. Importantly this ligand was more potent (lower doses) in neuropathic pain; representing an area of significant therapeutic need. In animal models there was no ventilatory depression and tolerance developed very slowly. In humans cebranopadol showed efficacy in chronic low back pain, has low abuse potential and respiratory advantage. The CORAL phase III trial in cancer-related chronic pain compared cebranopadol with morphine; cebranopadol was non-inferior. In a longer term safety and efficacy trial, CORAL-XT reported cebranopadol to be safe and well tolerated in prolonged treatment.
From this description of multi-targeting offering adverse effect advantage, non-selectivity is clearly being advocated. This goes against a large part of pharmacological dogma that drives selectivity to reduce adverse effect profile. With respect to opioids a rethink is needed; current development has already moved on (Fig 2).
Biased agonism
The fundamental principle governing biased agonism is that a particular ligand drives one signalling pathway over another and this produces therapeutic advantage. In the case of opioids, β-arrestin-2 KO animals display good quality antinociception but with reduced tolerance and other adverse effects. This has been questioned. The Trevena pharmaceutical company described a MOP receptor biased agonist; TRV130 or oliceridine that drove the Gi pathway over β-arrestin-2 recruitment, behaving as a G protein biased agonist. Based on these data it was presumed that it would have a reduced adverse effect profile. This was the case in preclinical studies and there was evidence of a similar effect in larger phase III clinical trials. These trials; APOLLO-1 (hard tissue, bunionectomy), APOLLO-2 (soft tissue, abdominoplasty) and Athena (safety and efficacy) facilitated Food and Drug Administration (FDA) approval of TRV130 as Olinvyk. There is documented respiratory advantage in humans. However, in a comprehensive series of experiments across several laboratories this biased agonist consistently returns as a partial agonist. G protein-coupled receptors support signal amplification. In the context of TRV130 if the G protein pathway is amplified and the arrestin pathway is not then a partial agonist could return a response at the former and not the latter; showing ‘apparent’ bias and the potential to misclassify. There is much debate in the literature covered above. From a therapeutic perspective this does not really matter as Olinvyk has some advantage over more conventional ‘non-biased’ ligands. Alongside TRV130 there are other G protein biased agonists such as PZM21. This is not in clinical development but is also returning as a partial agonist (Fig 2). In a very recent study Zhuang and colleagues used pharmacological-structural analysis to probe opioid interaction with MOP receptors. The MOP receptor is a GPCR and so composed of seven transmembrane (TM) domains; two areas were investigated, TM-3 face and TM-6/7 face of the ligand binding pocket. Importantly, TM-6/7 is involved in arrestin recruitment. Unbiased ligands (such as morphine and fentanyl) interact at both sites whereas putative biased agonists such as oliceridine and PZM21 preferentially interacted with TM-3.
Allosteric modulators
As noted, opioids bind to the orthosteric site on the receptor to engage G protein and produce an output. There are additional binding sites on the opioid receptor to which other molecules can bind and these can modify the activity of drugs binding to the traditional orthosteric site; these are allosteric modulators. Allosteric modulators can be positive, negative or neutral (silent) but I will focus on positive allosteric modulators (PAMs) here. Consider two situations; (i) endogenous opioid action and (ii) therapeutic intravenous opioid administration. The former is highly selective in site of action and temporal profile but the latter is not, with widespread systemic distribution. A PAM (alone not effective) has the potential to enhance selective endogenous opioid action thereby either reducing the need for a systemic approach or reducing the systemic dose; the net effect is retention of analgesia but with reduced adverse effect profile. The PAM enhances the natural temporal profile produced, in this example by the endogenous opioid ligand. Interestingly, allosteric modulator effects depend on the orthosteric ligand being used; this is called probe dependence. Allosterism concepts are covered in detail in excellent reviews. There are molecules in various stages of development that target opioid receptors. There are a significant amount of data on the BMS (Bristol-Myers Squib) series of allosteric modulators where positive variants (BMS-986122) produce good quality antinociception (mice and rats) against noxious heat and inflammatory stimuli with reduced ventilatory depression, constipation and reward. The orthosteric site was engaged by endogenous (produced or enhanced with an enkephalinase inhibitor) opioids, methadone or morphine. A similar set of data has been produced (also in mice) using MS1, a known MOP–PAM and a molecule with similar chemistry identified from a commercial database (Fig 2).
Opioid receptors, pain and epigenetics
In this review I have covered ligand regulation of receptor function and this makes sense from a pharmacological-therapeutic perspective. Opioid receptor expression and function can also be regulated by epigenetic mechanisms and if these changes can then be manipulated pharmacologically an extra layer to receptor regulation exists. According to the US National Institutes of Health National Human Genome Research Institute, epigenetics is ‘a field of study focused on changes in DNA that do not involve alterations to the underlying sequence’. Epigenetic regulation involves; histone modification, DNA methylation (adding a methyl group to cytosine in DNA) and the activity of non-coding RNAs (ncRNAs are RNA molecules that are not translated into protein). Epigenetic regulation is driven by a family of writers (adding modifications), readers (recognising) and erasers (removing modifications). Epigenetic mechanisms are implicated in a range of diseases including cancer (covered in a journal special issue), asthma and multiple sclerosis and epigenetic changes can be driven by environmental exposure, diet and age. Of relevance to anaesthesia, epigenetics has a role to play in the perioperative period: there are data showing epigenetic links to several types of pain and also opioid addiction-misuse which may have implications for the opioid misuse crisis.
There is thorough recent coverage of epigenetic control of opioid receptors by Reid and colleagues and others, much of this is based on in vitro experiments. Some of the issues relevant to MOP as a mainstay therapeutic opioid target are covered below.
It is generally accepted that in opioid use disorder (addiction) there is hypermethylation of MOP receptor promoters (where transcription is initiated). What are the effects of shorter-term therapeutic use? This has been addressed by Sandoval-Sierra and colleagues who examined genome-wide DNA methylation in the MOP promoter. Thirty-three opioid-naive dental surgery patients were recruited and provided saliva samples pre-surgery then 2.7 [1.5] and 39 [10] days post-surgery. There was a demonstrable hypermethylation of the MOP promoter confirming epigenetic regulation with short-term therapeutic opioid use; this was a small study. If opioid misuse starts with therapeutic use then DNA hypermethylation can be thought of as a continuum; starting in the clinic then continuing with inappropriate or illicit use in the community. DNA methylation of MOP promoters results in reduced expression in the brain.
Histones can be modified by acetyltransferases which add acetyl groups and Histone deacetylase (HDAC) remove them. Enhanced histone acetylation is reported in heroin users and there was a positive correlation with use history. HDACs have a role in several neuropathic pain syndromes where they are generally upregulated resulting in reduced histone acetylation. A study in rat bone cancer pain showed that the associated hyperalgesia is attenuated by the HDAC inhibitor trichostatin A potentially restoring acetylation. In the spinal cord of these animals, bone cancer reduced the expression of MOP and this was restored by the HDAC inhibitor. The authors also showed in vitro in PC12 cells that trichostatin A increased MOP messenger RNA (mRNA) and receptor protein. In a rat model of pancreatitis pain HDAC2 expression was also increased and MOP activity in the dorsal horn of the spinal cord was reduced; the HDAC inhibitor AR-42 attenuated this effect on MOP receptor immunoreactivity. Ricolinostat (an HDAC inhibitor) is currently in phase II evaluation for painful diabetic neuropathy where the investigators are examining pain intensity; there are no results posted (NCT03176472).
An additional mechanism by which opioid receptor expression can be regulated is via the action of non-coding RNAs; these generally target specific mRNAs to effectively silence expression, the best known being microRNA (miRNA). In tolerance (in vivo and in vitro paradigms) the miRNA let-7 inhibits MOP translation; others are variously involved. In a recent systematic review Polli and colleagues explored miRNA in human pains. They report that a wide range of types of pain; complex regional pain syndrome, fibromyalgia, migraine, irritable bowel syndrome, musculoskeletal pain, osteoarthritis and neuropathic pain, all have miRNA associations. We have considered HDACs above and there are data from other neurological diseases showing miRNA regulation of HDAC expression underscoring that epigenetic mechanisms should not be considered in isolation.
Long non-coding RNAs (lncRNAs) are involved in RNA stabilisation (including miRNAs) and the function of translated proteins involved in setting pain syndromes and in substance misuse. For example, an association with peripheral neuropathic pain, diabetic neuropathic pain, trigeminal neuralgia, central pain, inflammatory and cancer pain have been reviewed. Moreover, Michelhaugh and colleagues used Affymetrix microarrays to track five lncRNAs in the nucleus accumbens and these were upregulated in heroin users.
Is there a causal link between epigenetic modification and opioid receptor expression in the brains of patients with opioid use disorder (and as part of the continuum in early therapeutic use) and can this explain opioid misuse? In 1994 Gabilondo and colleagues measured opioid radioligand binding to post-mortem brain tissue. Density in the frontal cortex, thalamus and caudate nucleus in heroin users was similar to that in controls. Ferrer-Alcon examined post-mortem opioid receptor density (immuno technique) in the frontal cortex of patients with opioid use disorder and controls. They showed a reduction of MOP receptor (∼25%) in brains of users. Using positron emission tomography (PET) and [11C]diprenorphine, Williams and colleagues explored opioid receptor binding in living brains during early abstinence; they reported increased binding. If during addiction receptor numbers decrease, then in early abstinence it is not unreasonable to suggest a compensatory upregulation and an increase in [11C]diprenorphine binding. To address the epigenetic issue more directly Knothe and colleagues measured MOP expression by mRNA and protein along with DNA methylation in 27 post-mortem brains. They showed a good correlation between receptor mRNA and protein but not with DNA methylation. In a series of in vitro experiments that they ran concurrently, DNA methylation was an order of magnitude greater—highlighting differences between systems. The authors suggest that epigenetic mechanisms (addiction-related hypermethylation) are unlikely to control expression in the human brain. Epigenetic modulation of neurobiological circuitry is an attractive area of enquiry. Pharmacological manipulation of these epigenetic processes (pharmacoepigenetics) has the potential to affect responses.
Opioids—opioid receptors and immunomodulation
The fact that opioids modulate the immune response has been known for many years and we and others have reviewed this in the past. It is worth re-emphasising that immune modulation is not the same for all opioids. Moreover, there are recent data suggesting an interaction between opioids, COVID infection and outcome. The precise target site for immunomodulation is controversial with three areas of interest; none fully explains immune modulation. These are (i) the immune cell itself, (ii) modulation of the hypothalamic–pituitary–adrenal (HPA) axis and (iii) central actions. Taking these in reverse order, reactive gliosis in central pain is documented as are opioid receptors on glia and minocycline (microglial inhibitor) is effective in neuropathic pain. HPA axis modulation (increased glucocorticoids) appears to show marked species variation along with variation relating to acute or chronic administration. Direct modulation of the immune cell is the most controversial where there is evidence for effects of opioids on immune cell function. In a series of polymerase chain reaction (PCR) experiments we have failed to detect gene expression (mRNA) for any of the classical opioid receptors in mixed or separated human immune cell populations. Without mRNA then there can be no protein. In contrast we have detected mRNA for NOP and, in some cell types, its endogenous ligand N/OFQ. Moreover, we have recently used a novel fluorescent probe for NOP to detect active receptor protein and linked this to cellular function.
Toll-like receptors 4 (TLR4s) respond to the products of Gram-negative bacteria and are important in immune signalling. These receptors are widely distributed and can be found on the vascular endothelium and in a wide range of tumour cells. This receptor seems an unlikely target to be discussing when considering opioids and immune modulation but there is substantial evidence showing a range of opioids interact with this receptor and in a naloxone-sensitive manner. In the absence of definitive evidence for opioid receptor protein on immune cells, TLR4 is a plausible surrogate that can explain immune modulation. In the search for opioid receptor-mediated immunomodulation have we been looking in the wrong place?
Recent work from our laboratory has focussed on examining N/OFQ release from human polymorphonuclear (PMN) cells. We have developed a novel bioassay to measure the interaction of released N/OFQ with chimeric NOP receptors expressed in a biosensor layer of Chinese hamster ovary (CHO) cells. These receptors are forced to couple to Gαi/q proteins which, when activated, lead to measurable calcium responses. When polymorphs are overlaid onto sensor CHO cells and stimulated to degranulate with N-formyl-L-methionyl-L-leucyl-phenylalanine (fMLP) we can detect real time N/OFQ release from single cells. In unpublished observations we have reported similar responses in isolated human B and T cells.
Collectively, it is clear that NOP activation can modulate immune function and immune cells can produce and release N/OFQ. For classical opioids we should look elsewhere; TLR4 is a compelling target. Immune cells are also capable of the production and release of a range of opioid peptides for classical opioid receptors and these can then interact with neuronal opioid receptors producing a neuro-immune axis, and as discussed below for NOP–N/OFQ, an immune-vascular axis.
Sepsis and an immune-vascular axis; involvement of the N/OFQ opioid receptor
Sepsis is ‘a life-threatening organ dysfunction caused by a dysregulated host immune response to infection’. According to the UK sepsis trust there are 245,000 cases of sepsis each year, while 40% of sepsis survivors suffer permanent, life-changing after-effects and five people die with sepsis every hour. The dysregulated host response in sepsis and septic shock has profound effects on the cardiovascular system resulting in hypotension, organ hypoperfusion and resultant dysfunction/failure. This suggests cross-talk between immune and cardiovascular systems; an immune–vascular axis.
In a retrospective study we measured N/OFQ concentrations in critically ill patients in the ICU and compared these with their own recovery data and a matched control group of volunteers. We reported increased plasma N/OFQ concentrations over the first two days of admission to the ICU. Animal models have explored survival outcomes. In rats with caecal ligation and puncture peritoneal sepsis, mortality increased if N/OFQ was administered. Conversely, mortality was improved if the NOP antagonist UFP-101 was administered. The implications of these data are that N/OFQ is increased in sepsis (agrees with human ICU data) and that NOP antagonism might be beneficial as an adjunct to treat sepsis-induced hypotension. Indeed, in the rat microcirculation in vivo, we have shown that N/OFQ produced hypotension, vasodilation and macromolecular leak which was reversed by the NOP antagonist UFP-101.
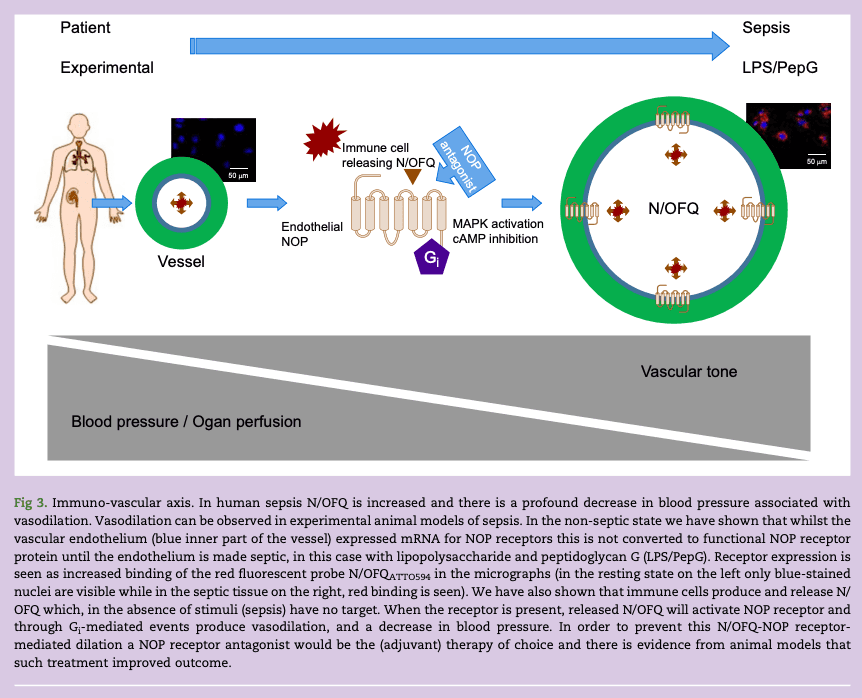
In a recent study we examined the expression of NOP on human vascular endothelium cells (HUVECs) and vascular smooth muscle. We showed that unstimulated endothelium expressed mRNA for the NOP receptor but expression of functional protein required treatment with lipopolysaccharide and peptidoglycan G (LPS/PepG) as an in vitro sepsis mimic. These upregulated NOP receptors are functionally active. In sepsis we hypothesise that immune cells release N/OFQ and that this then activates upregulated NOP receptors on the endothelium to support vasodilation and the sequelae of reduced blood pressure; this is an immune-vascular axis (Fig 3).
Figure 3.
Opioid-free analgesia
One school of thought to avoid opioid adverse effects (therapeutic and societal) is to eliminate them with the use of opioid-free analgesia. This seems a rather drastic course of action for a drug class that apparently has good efficacy when used for the right indication, at the right time and for the correct duration. There is literature exploring personalisation of opioid-free anaesthesia; or at least posing the question. There is a wide literature base exploring opioid-free techniques per se and a critical review recently compared opioid and opioid-free approaches concluding that ‘The data indicate that opioid-free strategies, however noble in their cause, do not fully acknowledge the limitations and gaps within the existing evidence and clinical practice considerations.’
Opioid free is worthy of some further consideration here; no opioids at all, no intraoperative opioids, no postoperative opioids or combinations? Olausson and colleagues performed a systematic review and meta-analysis of RCTs published between 2000 and 2021 looking at opioid-free general anaesthesia. The data were derived from 26 trials of 1,934 patients and concluded that opioid-free anaesthesia reduced postoperative adverse effects. Opioids were used in the postoperative period but their use was significantly lower in the opioid-free anaesthesia group. Fiore and colleagues also performed a systematic review and meta-analysis of opioid and opioid-free analgesia after surgical discharge. Their data were derived from 47 trials published after 1 January 1990 and which enrolled 6,607 patients. Opioid prescribing (compared with opioid free) did not reduce postoperative pain although the authors noted that ‘Data were largely derived from low-quality trials’. So what is the optimum? Reduced opioid multimodal intraoperative analgesia followed by limited postoperative (multimodal) use and transition to opioid free? Is this optimal for all procedures? There is guidance.
If this is all considered in the context of poor opioid stewardship (clinical use/prescribing) and the resulting well documented ‘opioid crisis’ then the scene is set for a potential withdrawal of opioids from routine use. This will deny a large patient population access to effective acute pain medications and in the case of cheap generics, effective analgesia in countries with developing health provision. In their report ‘Alleviating the access abyss in palliative care and pain relief—an imperative of universal health coverage: the Lancet Commission report’ Knaul and colleagues state of the 298.5 metric tonnes of morphine-equivalent opioids, 0.03% are distributed to low income countries. Moreover, using Haiti as an index country, the same report concludes 99% of (opioid) need goes unmet.
Conclusions
As pharmacologists and perioperative clinicians we have much to offer in the design of safer opioids (earlier part of this review), but the global community of researchers and perioperative practitioners need to tread carefully. Opioid medicine withdrawal might fix one important and perceived problem but then create another of monstrous proportions. What is clear is that the opioid epidemic has created a ‘hostile’ opioid environment; this hostility is not just from regulators but also from wider society and lawmakers. Whatever mechanisms underlie the actions of opioids and on the backdrop of the opioid crisis, it is clear that the route to market for any new opioid-based analgesics will not be straightforward; we must not give up.