Abstract
Opioid Use Disorder (OUD) is a chronic relapsing clinical condition with tremendous morbidity and mortality that frequently persists, despite treatment, due to an individual's underlying psychological, neurobiological, and genetic vulnerabilities. Evidence suggests that these vulnerabilities may have neurochemical, cellular, and molecular bases. Key neuroplastic events within the mesocorticolimbic system that emerge through chronic exposure to opioids may have a determinative influence on behavioral symptoms associated with OUD. In particular, structural and functional alterations in the dendritic spines of medium spiny neurons (MSNs) within the nucleus accumbens (NAc) and its dopaminergic projections from the ventral tegmental area (VTA) are believed to facilitate these behavioral sequelae. Additionally, glutamatergic neurons from the prefrontal cortex, the basolateral amygdala, the hippocampus, and the thalamus project to these same MSNs, providing an enriched target for synaptic plasticity. Here, we review literature related to neuroadaptations in NAc MSNs from dopaminergic and glutamatergic pathways in OUD. We also describe new findings related to transcriptional, epigenetic, and molecular mechanisms in MSN plasticity in the different stages of OUD.
1. Introduction
Medical use of opioids usually involves pain management, cough suppression, and antidiarrheal treatment, while psychiatric use includes opioid substitution of methadone and buprenorphine for the treatment of Opioid Use Disorder (OUD). Opioids have a high potential for abuse because they produce highly rewarding effects that can result in addictive involvement and frequent relapse following periods of abstinence (Koob et al., 2014; Schuckit, 2016). Many individuals begin their opioid use with prescription pain medications and subsequently require increased doses to reduce their symptoms, due to tolerance, or reduced behavioral effects with chronic dosing. In some cases, individuals can either no longer afford their prescription pain medications, or their prescriptions are discontinued, and then they switch to less costly, illicit opiates (e.g., heroin) (Kuehn, 2013). Opioid overdoses have become a nationwide epidemic within the United States (U.S.). The Center for Disease Control and Prevention (CDC) reported that the prevalence of fatal opioid overdose has steadily risen across the nation at an alarming rate. Since 1999, opioid-related overdose deaths have more than quadrupled in the U.S. (CDC, 2016). With the addition of the potent synthetic opioid, fentanyl, in heroin, a single opioid use can be fatal. The likelihood of a fatal drug overdose is greatly increased among those who have developed an OUD.
OUD is a clinical condition that, according to the 5th edition of the Diagnostic and Statistical Manual of Mental Disorders 5th ed. (DSM-V) of the American Psychiatric Association (2013), is characterized by compulsive use of opioids, drug craving, role dysfunction due to recurrent opioid use, drug use in physically hazardous situations, tolerance, withdrawal syndrome, repeated relapse, and other features. One of the hallmarks of OUD is that there is a long-term likelihood of relapse, despite treatment, due to an individual’s underlying vulnerabilities to this disorder (Schuckit, 2016).
There is evidence that the underlying neurobiological vulnerability for an OUD consists, in part, of persistent drug-induced structural and functional synaptic abnormalities at the level of the mesocorticolimbic dopamine (DA) system, which is widely considered to be essential for translating motivations into goal-directed action (Luscher and Malenka, 2011). Within this system, the nucleus accumbens (NAc) functions critically to integrate a variety of motivational input, and, under normal conditions, this structure mediates the processing of natural rewards relevant to survival (e.g., food and sex). Evidence suggests, however, that chronic exposure to certain unnatural rewards (e.g., drugs of abuse) can result in distinctive morphological alterations at the cellular level, along with corresponding neurophysiological changes (e.g., alterations in excitatory, glutamatergic and dopaminergic synaptic function, along with epigenetic and transcriptional alterations), that are associated with addiction-related behavioral adaptations, as well as the reorganization of neural circuits. In other words, drugs of abuse (e.g., psychostimulants, opioids) induce changes in key areas of the brain that make individuals more susceptible to compulsive use, conditioned responding to drug cues and contexts, and multiple relapses. Future treatment strategies for OUD, therefore, may be aimed at targeting these structural and functional adaptations in key brain regions.
In this review, we summarize the evidence for the functional and structural plasticity of dendritic spines of medium spiny neurons (MSNs) in the NAc and corresponding changes in neuronal connectivity and function that occur throughout the mesocorticolimbic system. We begin by providing context for our summary through a brief overview of the neuroanatomical and neurophysiological elements that influence opioid-induced structural and functional plasticity. Next, we summarize research related to the structural and functional plasticity induced by different stages of OUD, much of which has come from nonhuman animal models of addiction. We conclude by discussing possible ramifications of the findings for potential prevention and treatment strategies. Because it would be too large a task, our objective is not to provide a comprehensive review of the literature related to all of the plasticity mechanisms that come to bear upon each of the stages of OUD. Rather, we aim to elucidate some of the neurobiological underpinnings of OUD by describing examples of the relevant findings in hopes that future clinical and public health interventions may be devised.
2. Structural and functional plasticity of dendritic spines of medium spiny neurons in the nucleus accumbens
In this section, we provide context for our review of the literature related to opioid-induced plasticity of the NAc by briefly describing the structure and function of this region, in general, before directing more specific attention to its constituent neurons in addition to the plasticity mechanisms and processes that manifest at the level of MSN dendritic spines.
2.1. Nucleus accumbens
The rationale for investigating the role of the NAc in the facilitation of addictive behaviors comes, in part, from nonhuman animal-based evidence demonstrating its critical function in the integration of emotional and motivational processes (Floresco, 2015; Salgado and Kaplitt, 2015). In addition to mediating survival-related drives, such as feeding (Kelley et al., 2005) and sexual motivation (Everitt, 1990), the NAc has been shown to play a role in reinforcement learning (Everitt et al., 1991), impulsivity (Basar et al., 2010), and reward processing (Robbins et al., 1989). More relevantly, there is an abundance of evidence indicating that drugs with a high potential for reward exert their powerful behavioral effects largely through their action within the mesocorticolimbic DA system in general, and on the ventral tegmental area (VTA) and NAc, in particular (Luscher and Malenka, 2011; Thomas and Malenka, 2003). For example, nonhuman animal model research shows that opioids induce their rewarding influence by binding to mu opioid receptors in the VTA, which disinhibits dopaminergic neuronal firing, resulting in increased release of DA in the NAc (Johnson and North, 1992; van der Kooy et al., 1982). Additionally, in human neuroimaging studies, the functional connectivity between the NAc and other brain regions also has been shown to be altered in patients with OUD (Upadhyay et al., 2010; Zou et al., 2015).
The NAc is unique in its anatomical composition in that it consists of two functionally distinct subdivisions: a core and a shell. The core, which is part of the striatopallidal system, is involved in associative learning and conditioned responses (Parkinson et al., 2000), responses to motivational stimuli (Parkinson et al., 1999), and impulsive choices (Cardinal and Cheung, 2005). The shell, a substructure of the extended amygdala, has been shown to mediate the reinforcing effects of novelty (Parkinson et al., 1999), as well as substances with a high potential for reward, such as opioids (Alderson et al., 2001). In addition to this core-shell subdivision, there is emerging evidence that the lateral and medial aspects of the accumbal shell can be further subdivided according to the functional role that they play in the facilitation of rewarding behaviors and the experience of aversion (Al-Hasani et al., 2015; de Jong et al., 2019; Klawonn and Malenka, 2018; Lammel et al., 2014).
2.2. Medium spiny neurons in the nucleus accumbens
Both the core and the shell of the NAc consist primarily of MSNs, which are a variety of GABAergic inhibitory cells that may be categorized according to two distinct subtypes: those that project directly to the basal ganglia output nuclei, expressing primarily DA D1 subtype (DRD1) receptors, and those that project indirectly to the basal ganglia output nuclei and subthalamic nuclei, expressing DA D2 subtype (DRD2) receptors (Gerfen and Surmeier, 2011; Surmeier et al., 2007; Yager et al., 2015). A subpopulation of MSNs exhibiting both DRD1 and DRD2 also has been identified within the shell (Yager et al., 2015).
The MSNs of the NAc receive inputs from a variety of different cortical and subcortical areas, including the prefrontal cortex (PFC), the basolateral amygdala (BLA), the ventral hippocampus, the midline intralaminar thalamic nuclei, and the VTA. The NAc core receives glutamatergic projections from the prelimbic cortex and the BLA, whereas the shell is more densely innervated by glutamatergic afferents from the infralimbic cortex, ventral hippocampus, thalamus, and periventricular nucleus (Gipson et al., 2014; Kaplan et al., 2011a). The NAc shell also receives an abundance of DA projections from the VTA, while the core receives more scattered DA projections (Yao et al., 2008). These glutamatergic and dopaminergic projections synapse onto the dendrites of the MSNs. More specifically, and most frequently, they synapse onto tiny thorn-like protrusions on the dendrites, known as “dendritic spines” (Ethell and Pasquale, 2005).
2.3. Dendritic spines on medium spiny neurons
In mammalian brains, dendritic spines function as the primary postsynaptic interface from axons of most excitatory synapses (Bourne and Harris, 2008; Hotulainen and Hoogenraad, 2010; Robinson and Kolb, 2004; Spiga et al., 2014). Approximately 90 % of the 1014 synaptic connections in the brain terminate onto these subcellular structures, which range from ∼0.5 μm in diameter and 0.5–2.0 μm in length (Nimchinsky et al., 2002; Williams and Herrup, 1988). These chemical synapses transduce signals into electrical information that is transmitted throughout the neuronal circuit to the postsynaptic dendritic areas from the presynaptic axon terminals, and also molecular signals in the cell soma (Hotulainen and Hoogenraad, 2010).
Spines are composed of an actin cytoskeleton, vary according to size and shape, and consist of a bulbous head attached by a thinner neck, depending on their age. Glutamatergic inputs synapse onto the heads of spines and dopaminergic inputs synapse more distally onto the neck (Robinson and Kolb, 2004). “Synaptic triads” include GABAergic MSNs that receive axons from both dopaminergic and glutamatergic projections (Gipson et al., 2014; Spiga et al., 2014). The surface area of the head determines the strength of synaptic connections and differentiates spines into four main categories: stubby, thin, mushroom (Hotulainen and Hoogenraad, 2010; Peters and Kaiserman-Abramof, 1970), and filopodia (Fiala et al., 1998; Sorra and Harris, 2000; Spiga et al., 2014). Spine heads consist of a specialized organization of receptors, including glutamate (Glu) receptors, and hundreds of distinct supporting proteins, which compose the postsynaptic density (Yamauchi, 2002). In addition to organizing an assortment of signaling molecules at the postsynaptic membrane, the postsynaptic density (PSD) provides the structural framework for arranging neurotransmitter receptors, adhesion molecules, and ion channels (Hotulainen and Hoogenraad, 2010; Kennedy et al., 2005; Matus, 2000; Spiga et al., 2014).
3. Structural and functional plasticity of dendritic spines on medium spiny neurons
Structural plasticity refers to the process of change in size, count, morphology, and/or arborization of dendritic spines that develops through ordinary experience and learning or through exposure to various substances (Robinson and Kolb, 2004; Russo et al., 2010), particularly those that act upon dopaminergic and glutamatergic pathways, such as antidepressants, antipsychotics (Salgado and Kaplitt, 2015), and prototypical drugs of abuse (e.g., psychostimulants, ethanol, opioids, etc.). These structural changes, which can persist for months, are modulated largely by the rearrangement of the actin cytoskeleton (Hotulainen and Hoogenraad, 2010). Drugs of abuse can have a direct influence on the rearrangement of the actin cytoskeleton. Numerous genes encoding for cytoskeleton regulatory proteins are influenced by opioid and stimulant drugs. For example, two scaffolding proteins related to the postsynaptic cytoskeleton, PSD-95, and Homer 1, are decreased by exposure of the NAc to both morphine (Spijker et al., 2004) and to cocaine (Heiman et al., 2008; Roche, 2004; Szumlinski et al., 2006; Yao et al., 2004). Moreover, both morphine (Spijker et al., 2004) and cocaine (Kim et al., 2009) reduce GTPases that are involved in regulating the actin cytoskeleton (i.e., RhoA, Rac1, and cell division cycle 42 (Cdc42)) (Russo et al., 2010). Opioids activate transcriptional regulators in the NAc, such as the transcription factors DeltaFosB and cyclic adenosine monophosphate response element binding protein (CREB) (Alibhai et al., 2007; Hope et al., 1994; Shaw-Lutchman et al., 2002, 2003) and have similar effects on the genes that regulate cytoskeleton arrangement and other genes. Approximately 25 % of all genes associated with structural and synaptic plasticity, such as activity-regulated cytoskeletal protein, actin-related protein-4, and cofilin (McClung and Nestler, 2003; Renthal et al., 2009) are attributable to DeltaFosB (Russo et al., 2010), which is also implicated in drug-induced alterations of the density of dendritic spines (Maze et al., 2010). Although drug-induced changes to spine density can emerge, even in the absence of functional changes to the cell, through the creation of new silent synapses, or by the consolidation of existing synapses into stronger ones, functional changes can occur by means of drug-induced changes to the spine size or shape, though not necessarily the number of spines (Russo et al., 2010).
Synaptic plasticity refers to the ability of synapses to increase or decrease in strength or function, also in accordance with experience, or as a consequence of exposure to various substances (Kauer and Malenka, 2007). Because plasticity at NAc excitatory synapses was ostensibly selected (via natural selection) to serve some adaptive behavioral functions, such as the facilitation of learning and memory, it is widely understood that drugs of abuse “hijack” mesocorticolimbic DA circuitry via multiple mechanisms of synaptic plasticity, resulting in the long-lasting pathological behaviors associated with addiction (Hyman and Malenka, 2001; Hyman et al., 2006; Kalivas and Volkow, 2005; Kauer, 2004; Kelley, 2004; Montague et al., 2004). Synaptic plasticity depends upon lasting increases in synaptic strength (i.e., long-term potentiation (LTP)), as well as decreases in synaptic strength (i.e., long-term depression (LTD)).
Some of the molecular mechanisms underlying synaptic plasticity include the number of glutamatergic or Glu receptors (e.g., N-methyl-d-aspartate receptor (NMDAR) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR)) located at the synapse (Gerrow and Triller, 2010), the quantity of neurotransmitters present within the synapse, and how efficiently cells react to those neurotransmitters (Gaiarsa et al., 2002). Multiple types of synaptic plasticity are expressed by the excitatory synapses within the NAc, including LTP dependent upon endocannabinoid receptors, NMDAR-dependent LTD and LTP, as well as a presynaptic form of LTD generated by metabotropic Glu 2 subtype receptor (GluR2) (Luscher and Malenka, 2011). LTP within the NAc is characterized by the increase of AMPAR-mediated components of the synaptic reaction and a corresponding decrease in the NMDAR-mediated components (Kombian and Malenka, 1994; Thomas and Malenka, 2003). This describes the AMPAR:NMDAR ratio, a surrogate measure of synaptic efficacy. The addition or deletion of AMPARs or NMDARs, as well as alterations in the subtype composition of AMPARs, determines synaptic strength (Volkow and Morales, 2015). For example, the upregulation of GluR2, a high calcium permeable AMPAR subtype (Guire et al., 2008), influences cocaine-induced increases in AMPAR:NMDAR ratios related to LTP (Boudreau et al., 2007; Conrad et al., 2008; Kourrich et al., 2007), and enhances NAc MSN responsivity to Glu elicited by drugs or drug cue exposure (Volkow and Morales, 2015; Wolf and Ferrario, 2010). As we observe below, in addition to drug administration, contexts and cues associated with drug administration appear to play a crucial role in the facilitation of synaptic plasticity, an effect that is likely a consequence of the drug’s molecular actions in combination with the brain’s cue and contextual learning associated with the experience of the drug (Luscher and Malenka, 2011).
Structural and synaptic plasticity emerge in a dynamic, reciprocally reinforcing relationship with one another, whereby the strength of the synapse influences the size and shape of the spines, and the morphological attributes of spines influences the strength of synaptic connections (Robinson and Kolb, 2004). The electrical properties of neurons are influenced by the morphological attributes of dendrites and dendritic spines. For example, since action potential generation is affected as electrical current flows toward the soma through dendrites, which filter post-synaptic potentials, the electrical signaling may be affected by alterations in dendritic complexity (Segev, 2006). Because neurons are primarily connected by means of dendrites and dendritic spines (Kobrin et al., 2015), and there is evidence that the attenuation and development of these spines can result in the weakening or strengthening of synapses, respectively (Gipson et al., 2014), the various measures of dendritic spines have been considered important indices of synaptic plasticity. For instance, the development of new spines and enlargement of existing spines may result in some forms of LTP (Carlisle and Kennedy, 2005; Yuste and Bonhoeffer, 2001), whereas diminution and reduction of spines has been associated with LTD (Okamoto et al., 2004). LTP also can result in a more functional spine through the anchoring of AMPARs, whereas LTD can result in the degradation of spines (Bourne and Harris, 2007; Tada and Sheng, 2006). LTP and LTD, which can initiate alterations in signaling pathways, are believed to introduce changes in the production and constraint of cytoskeleton proteins, thus affecting spine development and stability by altering the polymerization of actin (Russo et al., 2010). Because they consist of mostly NMDAR, and little to no AMPAR (Malenka and Nicoll, 1997), silent synapses, which can be represented by an increase in thin spine and synaptic depression (Shen et al., 2009; Thomas et al., 2001), are ideal for long-term plasticity (Marie et al., 2005). There is evidence that the size of the spine head corresponds proportionally to its synaptic capacity, as well as the number of presynaptic docked vesicles and postsynaptic receptors (Carlisle and Kennedy, 2005). The characteristic mushroom shape of spines emerges upon their stabilization, along with larger postsynaptic densities (Harris et al., 1992) and increased AMPAR surface expression (Holtmaat et al., 2005; Zuo et al., 2005).
Molecular mechanisms in models of opioid addiction impact structural and functional plasticity, circuitry remodeling, neurophysiological changes in these circuits and result in addiction-related behaviors (Graziane et al., 2016). Graziane et al. (2016) utilized repeated morphine administration models for sensitization and conditioned place preference (CPP) in rodents and measured silent synapses in accumbal neurons. A silent synaptic contact between two neurons, which consists exclusively of NMDARs, occurs when presynaptic action potential fails to initiate postsynaptic signal. This repeated morphine administration paradigm generated accumbal silent synapses and decreased long-thin dendritic spines in MSNs. However, co-administration of the GluA2 peptide blocked AMPAR internalization and morphine-induced generation of silent synapses. Administration of this GluA2 peptide into the NAc prior to conditioning blocked drug preferences in a morphine CPP paradigm, an experimental technique used to assess the reinforcing effects of drugs. In addition to suggesting that NAc shell silent synapses induce the remodeling of circuits via GluA2 receptor internalization, which appear to be necessary for morphine reward learning, this study represents an example of research that incorporates molecular mechanisms of neural plasticity regulating neurophysiology, accumbal circuitry, and drug-related behaviors.
4. Epigenetic and transcription factor mechanisms for plasticity in the NAc
Opioids have several major mechanisms for producing rewarding and reinforcing effects. As noted previously, they bind to mu-opioid receptors in the VTA and produce disinhibition of DA neuronal firing, resulting in increased synaptic DA in the NAc (Di Chiara and Imperato, 1988; Johnson and North, 1992). Opioids also directly activate mu-opioid receptors on NAc neurons, and any disruption of these opioid-activated accumbal neurons disrupt opioid reinforcement responses (Zito et al., 1985). Opioid-induced downstream regulation of effectors produces changes in signaling cascades, transcriptional activity and activation, and epigenetic modifications, all of which result in the translation of new proteins that produce synaptic changes in the NAc and serve as the basis for new memories (Hyman et al., 2006).
Transcriptional activation is important in the regulation of neuroplasticity, dendritic plasticity, and ultimately addiction-related behaviors. For example, opioid receptor activation alters production of effector molecules that travel to the cell nucleus, where they interact with the transcription factor, CREB, resulting in gene transcription (Chartoff et al., 2009). CREB binds as a dimer to the cyclic AMP-response element (CRE) sites in the regulatory region of several genes, while certain cell surface receptor activations result in the phosphorylation of the CREB-binding protein (CBP), which assembles to produce a larger transcriptional complex. These complexes then modify the N-terminal tails of histones, proteins that are wound around DNA, which are then covalently modified and impact the interactions between the histone and DNA. Transcriptional complexes promote processes such as histone acetylation, which alter the conformation of the nearby chromatin (Horn and Peterson, 2002), and enable the synthesis of RNA by RNA polymerase II. Morphine treatment alters multiple transcriptional regulators, including histone acetyltransferases, deacetylases, methyltransferases, demethylases, DNA methyltransferases, oxidases or demethylases known as ten-eleven translocation proteins, all of which modify histones and chromatin conformation (Browne et al., 2020).
Signal transduction by opioids can lead to receptor and G protein effector activation and the downstream phosphorylation of CREB or other transcription factors that alters transcriptional machinery and enables the synthesis of new proteins that alter plasticity. Some studies have demonstrated that levels of CREB activity in the NAc can be regulated by either rewarding or aversive environmental stimuli, including morphine administration (Barrot et al., 2002). Using CRE-LacZ reporter mice, this last research group demonstrated that both morphine reward and stress activate CRE-mediated transcription in the NAc.
Epigenetic changes in neurons alter gene expression and not nucleotide sequence of DNA through conformational modification to chromatin structure and accessibility. Opioid addiction states can produce a more accessible chromatin state through these epigenetic processes allowing greater plasticity-related gene expression, resulting in accumbal synaptic and dendritic changes (Cahill et al., 2018). These modifications alter basal levels of gene transcription, which are important in neuronal function and plasticity. For example, in post-mortem striatum tissues from individuals with OUD, other epigenetic changes were found relevant. Hyperacetylation of lysine 27 on histone H3 in the striatum was correlated with heroin use history (Egervari et al., 2017). In this study, similar results were found in striatal tissues of heroin self-administrating rats. Epigenetic regulation of a histone methyltransferase was produced by chronic morphine and altered reward, sensitization (i.e., chronic, intermittent drug treatment that increases motor activity), drug withdrawal signs, and tolerance behaviors, all of which suggests novel chromatin-based mechanisms in opioid addiction (Sun et al., 2012). Repeated opioid treatment produces changes in the function of transcription factors and epigenetic factors which remodel chromatin structure and change DNA accessibility in opioid reward neuroplasticity.
Another transcription factor mediating responses to drugs of abuse is activator protein-1 (AP-1). AP-1 is composed of heterodimers of the Fos family which include c-Fos, FosB, Fra1, and Fra2, along with the Jun family of c-Jun, JunB, and JunD. These transcription factors are rapidly expressed after acute drug exposure (Browne et al., 2020). Acute morphine also induces short-term immediate-early genes of the c-Fos family gene in the NAc (Bontempi and Sharp, 1997; Leite-Morris et al., 2002). Though AP-1 activity is short-lived (i.e., hours), its effects can be lengthened by drug-induced expression of DeltaFosB proteins, which are modified isoforms of FosB that accumulate with repeated drug treatments and produce long-lasting behavioral and neuroplastic effects (Chen et al., 1997). Nestler’s group went on to utilize a DeltaFosB transgene in mice, which targeted a subgroup of striatal MSNs expressing enkephalin (Zachariou et al., 2006). In this study, accumbal DeltaFosB overexpression increased the sensitivity of the mice to morphine reward. Our group (author GBK) demonstrated that treatment with a repeated and intermittent morphine reward results in increased locomotor activity in a manner consistent with the development of drug sensitization. This intermittent morphine pre-treatment produced a significant induction of FosB/DeltaFosB in multiple brain regions, including prelimbic (PL) and infralimbic (IL) cortices, NAc core, dorsomedial caudate-putamen (CPU), basolateral amygdala (BLA), and central nucleus of the amygdala (CAN), but not in a motor cortex control region (Kaplan et al., 2011b). The FosB/DeltaFosB plasticity in these regions may contribute to the opiate-induced sensitization observed. Overexpression of DeltaFosB in the NAc has been shown to enhance morphine reward and signs of morphine withdrawal, in addition to altering morphine’s analgesic and tolerance effects (Zachariou et al., 2006). Thus, repeated opioid exposure induces DeltaFosB, which may mediate long-term, addiction-related transcriptional activity leading to epigenetic modifications.
The structural changes to NAc dendrites induced by exposure to drugs of abuse is determined by actin cycling pathways, which are regulated by transcription and epigenetic processes. For example, heroin self-administration in rats negatively regulates the actin-binding protein drebrin in the NAc. Debrin overexpression in the NAc decreases heroin seeking and increases dendritic spine density, whereas debrin knockdown enhances these effects. Drebrin expression is regulated by the transcriptional repression of the histone modifier, HDAC2 (Martin et al., 2019). Inhibition of actin polymerization in the NAc reduces morphine-induced CPP, an effect that was persistent after a single injection of the actin polymerization inhibitor and was not reversed by a morphine prime (Li et al., 2015).
In summary, the structural and synaptic plasticity of dendritic spines within the NAc depends on a variety of cellular and molecular mechanisms involving multiple signaling pathways, transcription factors, epigenetic processes and cytoskeletal proteins. In what follows, we consider the effects of different stages of OUD on the plasticity of accumbal spines and synapses and their mechanisms.
5. Opioid use disorder stage-dependent plasticity of NAc dendritic spines
From a behavioral standpoint, OUD can be characterized by impaired inhibitory control over the amount, frequency, and duration of involvement with opioids, despite adverse consequences, because of craving (American Society of Addiction Medicine, 2011). The addiction cycle can be characterized by two discrete behavioral stages: active involvement and abstinence. These addiction-related behaviors correspond to a range of affective experiences, which vary with respect to valence and intensity (see Fig. 1). The cycling of these affective experiences characterizes the various motivational influences underlying either involvement or abstinence. For example, early abstinence is frequently accompanied by the negative affective experiences of restlessness, irritability, and dysphoria, which tend to motivate a return to involvement through what has been referred to as “withdrawal relief craving” (Heinz et al., 2003). Notably, another type of relief commonly sought by addicted individuals, which is reflected through the experience of craving, emerges in the context of uncontrollable stress. Whether exogenous (e.g., due to environmental factors) or endogenous (e.g., due to the allostatic state that emerges through repeated involvement (Koob et al., 2014)), such stress has the capacity to increase the hedonic property of opiates, thereby hastening the progression from use to abuse to addiction. In line with this ‘self-medication hypothesis,’ prior research has revealed that intense, uncontrollable stress can “prime” the opioid system by enhancing the pharmacologic potency of both opioid receptor agonists and antagonists (Drugan and Maier, 1986; Williams et al., 1984), as well as the reinforcing potential of heroin (Stafford et al., 2019). Craving also may be initiated and perpetuated entirely by the positive affective states (e.g., tranquility, euphoria, etc.) that accompany opiate use, through what has been referred to as “reward craving” (Heinz et al., 2003).
Fig. 1. Opioid-Induced Plasticity of the Nucleus Accumbens in Relation to Addiction Behavior and Corresponding Affective Experience.
A schematic depiction of the cycle of addiction in relation to both behavior and affective experience. This figure represents the combination and adaptation of a diagram from Koob and Le Moal (2001) and a diagram by Russell and Feldman Barrett (1999). The circumplex at the center of the figure represents the affective experiences that tend to correspond to the two discrete behavioral phases of addiction: acute involvement with a substance or activity and acute/protracted abstinence. These affective experiences tend to vary with respect to valence (horizontal axis) and intensity (vertical axis), depending on the substance or activity of involvement, the duration of involvement, and the duration of abstinence. The arrows constituting the outer circle represent the cyclical nature of the addictive experience, which coincides with both behavior and affective states. The individual affective states that are indicated within the circumplex are not meant to be exhaustive, but rather representative of the types of states that characterize a given quadrant (e.g., high intensity, negative valence). The paradigms indicated beneath the circumplex refer to the types of experimental designs that correspond to either the involvement or abstinence phase of the addiction cycle. Abbreviations: SA = Self-administration; Sensi = Sensitization; CPP = Conditioned Place Preference; CPA = Conditioned Place Aversion. Beneath these experimental paradigms are the structural plasticity changes that that have been found in the preponderance of studies within the nucleus accumbens.
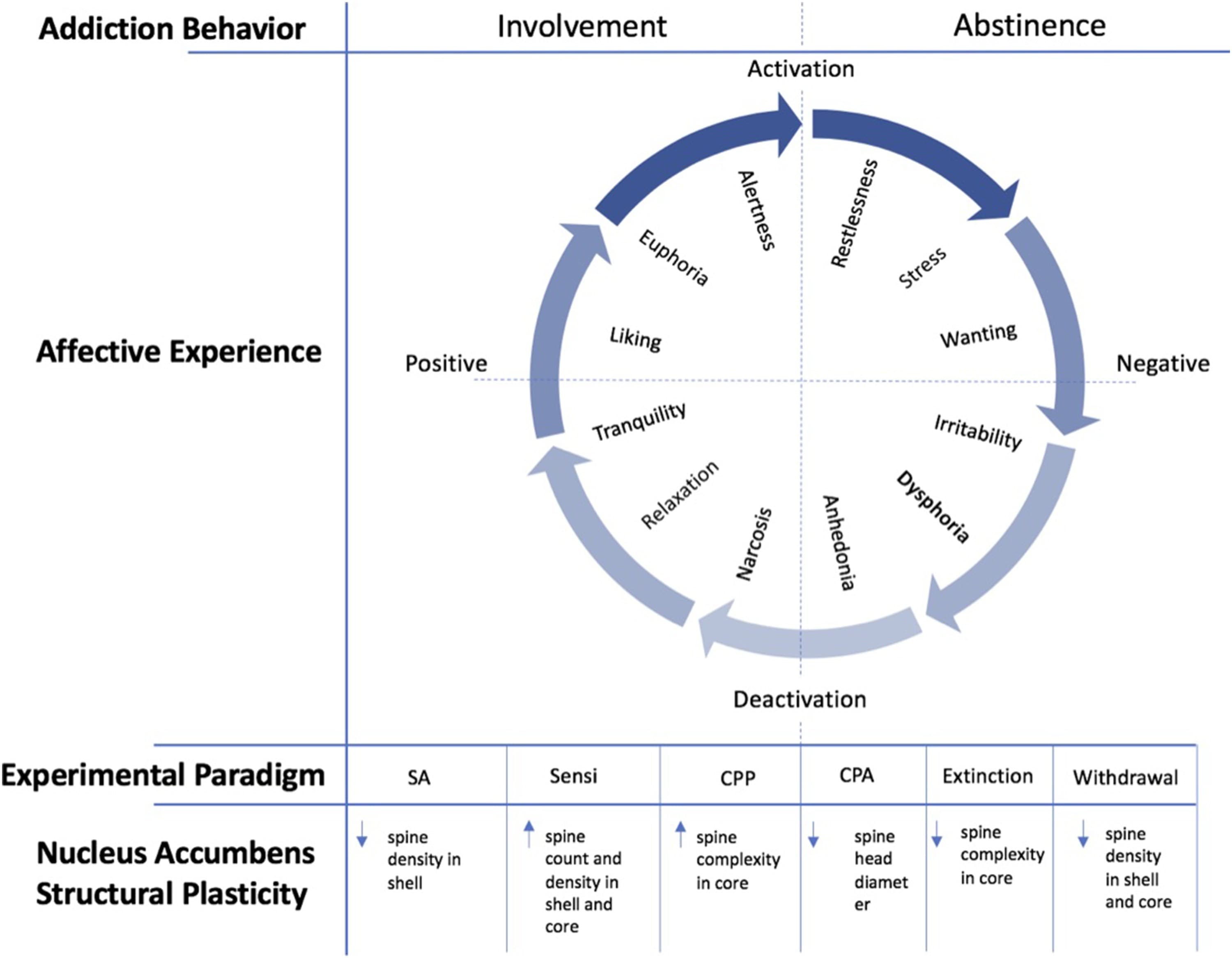
In this section, we review the evidence for opioid-related structural and synaptic plasticity of NAc MSN dendritic spines provided by nonhuman animal models of the stages of addiction within the conceptual framework provided above. We begin by describing the active involvement stage and the nonhuman animal models that represent it, such as the self-administration, sensitization, and CPP paradigms. Next, we discuss conditioned place aversion and somatic aversion paradigms as part of the acute abstinence phase of addiction. We then discuss drug craving and relapse (including drug prime and stress-induced reinstatement of reward and self-administration) as being representative of the protracted abstinence stage of addiction. In each section, we review the synaptic and structural plasticity associated with each stage of addiction. Finally, we consider models of treatment and recovery for OUD, including extinction of CPP and self-administration.
6. Opioid involvement and accumbal plasticity
The Active Involvement phase of OUD refers to acute intoxication with opioids. Initial exposure to a substance among humans can be influenced by a variety of factors, including social motives (e.g., the desire to conform to peer pressure) and emotional motives (e.g., the desire to escape negative emotions or amplify positive emotions). Although initial involvement with commonly abused drugs is frequently characterized by hedonia (e.g., the “hit” or “high”), or the experience of “liking” the substance (Berridge and Robinson, 2016), this experience is hypothesized to gradually diminish as an allostatic state emerges (Koob et al., 2014). Molecular mechanisms from single use of opioids come from gene expression studies. Even single doses of morphine (10 mg/kg) and heroin (10 mg/kg) induce gene expression of molecules involved in signal transduction, apoptosis, the cell cycle, enzyme inhibitor activity, the stress response and cell differentiation (Piechota et al., 2010). Single-dose morphine administration (15 mg/kg, s.c.) does not alter total dendritic spine densities in the NAc of wild-type mice. In contrast, the same morphine administration increased total dendritic spine densities in the NAc shell and core in CB-1 knockout mice, suggesting a modulatory role of CB-1 in acute morphine effects of structural plasticity in the NAc MSN (Guegan et al., 2016).
Human neuroimaging studies of acute and chronic opioid exposure have revealed important information regarding the effect these substances have on reward circuitry. However, these clinical studies are less able to provide detailed analysis of signal transduction pathways, neurophysiological changes, molecular and epigenetic changes, and structural plasticity changes than confocal microscopy does in nonhuman animal models. Therefore, several nonhuman animal models have been utilized to permit the analysis of these elements. We next discuss three paradigms that have been particularly revealing with respect to the effects of opioids on the structural and functional plasticity of the dendritic spines of NAc MSNs – namely, self-administration, sensitization, and conditioned place preference.
6.1. Models of active involvement in opioid use disorder
6.1.1. Opioid self-administration
Self-administration refers to the operant behavior of a human or nonhuman animal in response to the reinforcing efficacy of a drug (Schuster and Thompson, 1969). In this model, subjects are willing to work to achieve self-administration. This model of behavior rests on the premise that actions preceding positive reinforcement will be strengthened and more likely to be repeated. Drugs with a tendency to promote self-administration correspond well with those that are commonly abused among humans (Koob et al., 2014). In experiments employing a drug self-administration model, animals are trained to perform an operant response (e.g., bar presses or nose pokes) to receive an intravenous drug infusion (e.g., cocaine or morphine), which is paired with a distinct cue (e.g., tone or light) or a specific context (Kalivas, 2009; Wolf, 2016). The operant response can be manipulated by increasing the reliability, as well as the dose of drug administration. For example, in cocaine self-administration, animals tend to increase their rate of cocaine self-administration if the dose is decreased, whereas they tend to decrease their cocaine self-administration if the dose is increased (Koob et al., 2014). The drug self-administration paradigm is relevant to both the active involvement stage and the experience of craving (which is detailed later), inasmuch as it provides a measure of drug wanting, as indexed by the drug seeking behavior of the animal. Importantly, self-administration is differentiated from experimenter administration, a paradigm in which the experimenter administers the drug, irrespective of environmental contingencies. Although experimenter administration provides important information about the effects of the drug, it does not provide a measure modeling drug craving.
In self-administration models, long-term treatment with opioids have been shown to decrease the dendritic branching and spine density within the NAc and the prefrontal cortex (PFC). This occurs, whether morphine is administered through self-administration or experimenter administration (Robinson and Kolb, 2004). Although both self-administered and experimenter-administered morphine decreased dendritic spine density in the NAc shell, self-administration had a more substantial effect in this regard (Robinson et al., 2002). The effect of morphine on dendritic spine density persisted for at least a month, and the morphine-related effects on spinal density depended on both the brain region and mode of administration (Robinson et al., 2002). Moreover, self-administration has been shown to produce a differential effect on the morphology of dendritic spines compared to experimenter administration, which suggests that volition might be a relevant factor in the resultant plasticity (Russo et al., 2010). A key feature of brain reward circuitry plasticity in mediating drug seeking is increased Glu release into the NAc from many sources including the PFC, amygdala, hippocampus, thalamus, and activation of Glu-DA synaptic connections in MSNs. For example, reinstatement of heroin self-administration is produced by a heroin prime or a drug cue, resulting in increased extracellular glutamate in the NAc (LaLumiere and Kalivas, 2008). These alterations in the extracellular basal levels of Glu have been proposed to contribute to the reduction of MSN synaptic plasticity after heroin self-administration (Gipson et al., 2014; Peters et al., 2009). Self-administration of heroin induces persistent reductions of the glial Glu transporter GLT-1 in the NAc (Gipson et al., 2014; Shen et al., 2014), and GLT-1 protects these neurons from the excitotoxic action of Glu (Nakagawa and Satoh, 2004). There is also evidence that the surface expression of metabotropic Glu 1 subtype receptor (GluR1) in both the NAc shell and core may be attenuated through chronic self-administration of morphine (Glass et al., 2008). In the NAc shell, chronic morphine self-administration resulted in a reduction in surface GluR1 in DRD1-expressing MSNs, whereas, in the core, surface GluR1 was decreased in non-DRD1 receptor expressing MSNs. After chronic alternating injection of increasing doses of morphine, ultrastructural measures of plasticity of metabotropic GluR1 increase among MSNs of the NAc shell that respond to Glu- or DA-induced DRD1 stimulation (Glass et al., 2008). Additionally, in this paradigm, GluR1 receptors, which are responsive to Glu, but not DRD1 receptor activation, are enhanced in MSNs within the core. Changes in GluR1 expression, which are regulated, in part, by phosphorylation (Hakansson et al., 2006; Vinade and Dosemeci, 2000), can precipitate alterations in the morphology of dendritic spines (Kopec et al., 2007).
Another putative mechanism of plasticity in opioid self-administration involves gene expression of dendritic structural molecules in dendrites. Axon guidance molecules, such as integrins, semaphorins, and ephrins, produce neuroadaptations via axon-target connections and synaptogenesis and were found to be upregulated in oxycodone self-administration studies (Yuferov et al., 2018). For example, this last study found opioid self-administration induced two specific integrins and a semaphorin, Sema7a, in the NAc. They also demonstrated downregulation of one gene from the ephrin receptor family, Epha3, in NAc cells. These molecules are involved dendritic spine morphology via interactions with integrins. Another example of plasticity mechanisms in opioid self-administration suggests the involvement of micro-RNAs in neuroplasticity and axonal guidance in the NAc (Tapocik et al., 2013). MicroRNAs (miRNAs) are a class of small noncoding RNAs involved in the regulation of gene expression at the posttranscriptional level by degrading their target mRNAs of inhibiting their translation. In this last study, morphine self-administration-induced miRNA genes H 19, miR-675 and miR-154, which regulate mu-opioid receptors and DA neuron differentiation. These miRNAs are candidates to shape accumbal dendritic architecture by opioids in an activity-dependent manner.
6.1.2. Opioid sensitization
Sensitization is an associative learning process in which repeated or intermittent administration of a drug in a specific environment produces increased behavioral and locomotor effect of that drug as a consequence of past exposure (Brown et al., 2011; Kaplan et al., 2011b; Robinson and Kolb, 2004). Because sensitization can persist for days, weeks, or months, the behavioral changes that characterize addiction (e.g., drug-seeking, drug-taking, relapse) are thought to be mediated, in part, by this aberrant learning process, which emerges from key synaptic and structural changes that occur among motor, hedonic, and cognitive neural systems through chronic exposure to certain drugs (Alcantara et al., 2011; Kaplan et al., 2011b; Robinson and Berridge, 1993).
There is evidence that sensitization can be induced by the stimulant effects of opioids (Babbini and Davis, 1972; Powell and Holtzman, 2001; Vanderschuren and Kalivas, 2000), in addition to the conditioned rewarding effects of opioids (Kaplan et al., 2011b; Lett, 1989; Shippenberg et al., 1996). Structural changes in some parts of the NAc, but not the other, is associated with the development of sensitization (Robinson and Kolb, 2004). For example, increases in the ratio between neuron and asymmetric synapses – a characteristic feature of the glutamatergic synapses that emerge between the accumbens and cortical, amygdalar, hippocampal, and/or thalamic inputs – within the NAc shell, but not the core, have been shown to be associated with morphine-induced behavioral sensitization (Alcantara et al., 2011). However, behavioral sensitization induced by morphine has also been shown to be associated with increases in the count and density of dendritic spines within both the NAc shell and core (Guegan et al., 2016). A purported mechanism underlying this sensitization-related plasticity is the accumulation of FosB/DeltaFosB transcription factors within the NAc, among other components of the mesocorticolimbic system (e.g., prelimbic and infralimbic cortices, amygdala), which has been shown to be induced by morphine administration (Kaplan et al., 2011b).
Another mechanism for plasticity in accumbal neurons is opioid sensitization via gene changes in expression. For example, in conditioned morphine sensitization, 155 genes were upregulated and 88 were downregulated (Liang et al., 2011). Several gene transcripts were chosen to confirm changes via quantitative real-time polymerase chain reaction (qRT-PCR). These identified genes had functional involvement in receptor-ligand interactions, synapse plasticity, ion transport, and protein phosphorylation.
Gene expression and epigenetic studies have revealed other molecular mechanisms for opioid sensitization. For example, using a sensitization paradigm, Sun et al. (2012) observed that five to seven doses of repeated daily intraperitoneal morphine injections downregulated histone modification through a histone methyltransferase (H3K9me2), and this effect was dose-dependent. In this study, this reduced H3K9me2 binding followed glutamatergic signaling via three genes (grin2a, grm5, grm8), which may represent different pathways of gene expression. Another molecular mechanism for opioid sensitization-induced changes in dendritic plasticity involves the cannabinoid receptor subtype (CB1-R), the deletion of which blocks the development of morphine locomotor sensitization (Guegan et al., 2016). Guegan et al. (2016) demonstrated that dendritic spine density in the NAc shell and core in wild-type (WT) mice, but not controls, increased upon morphine injection one week following chronic morphine treatment. This increase in spine density in WT mice was significantly higher in the NAc core when compared with CB1-R knockout mice.
6.1.3. Opioid conditioned place preference
The incentive salience that emerges through mesocorticolimbic mechanisms describes the process whereby a previously neutral stimulus is imbued with positive motivational value through its association with a rewarding stimulus and elicits approach behavior (George and Koob, 2017; Robinson and Berridge, 1993). Exposure to drug-predictive cues stimulates the release of DA within key areas such as the NAc (Berridge and Robinson, 2016), PFC (Milella et al., 2016), and amygdala (Fotros et al., 2013), which results in drug rewards becoming disproportionately sought, relative to natural rewards (Berridge, 2007; Kelley and Berridge, 2002). As mentioned previously, opioids induce their distinctive reward as they disinhibit interneurons of the VTA, by binding onto their mu opioid receptors, thereby stimulating the release of DA into the NAc (Johnson and North, 1992). As opioid reward becomes paired with specific cues and contexts, these environmental stimuli are instilled with motivational significance via cortical glutamatergic afferents to the NAc (Pecina and Berridge, 2013). Subsequent exposure to these stimuli, even in the absence of drug reward, can induce DA transmission in the NAc core and activate DRD1 receptor expressing MSNs in the NAc core (Calipari et al., 2016; Ito et al., 2000), which has been proposed to precipitate craving for the conditioned reward (Berridge, 2007; Berridge and Robinson, 2016). This hyperdopaminergic state may give rise to the experience of reward craving.
The CPP paradigm is an experimental technique that has been used extensively in addiction research to assess learning in nonhuman animals; it measures the reinforcing effects of drugs by means of classical conditioning (Kobrin et al., 2017; Koob et al., 2014; Tzschentke, 2007). The CPP paradigm serves as a model for the phenomenon in humans whereby repeated drug use becomes paired with various cues and contexts in the environment that represent past associations, which elicit craving for future drug use and potentially relapse (Franken et al., 1999). Therefore, CPP models both single-dose conditioned and cued effects of drug reward. In a model of the CPP paradigm, animals are exposed to two distinctive contexts and/or cues that are paired with either drug administration or saline administration and then given the option to spend time in either context. When the animal spends more time in the drug-associated context than in the non-drug context, CPP is said to have developed. Because animals tend to show a preference for contexts that are associated with positive reinforcers (e.g., drugs of abuse), time spent in the drug-associated context, which is dose-dependent, is considered to be a measure of the presence and potency of the reinforcing effects of the drug (Kobrin et al., 2015; Koob et al., 2014).
CPP-related behavioral adaptations are accompanied by structural and synaptic adaptations of the dendritic spines of MSNs in the NAc. For example, there is evidence that morphine CPP is associated with enhancements in the dendritic complexity, including length and intersections, within the NAc core (Kobrin et al., 2015). (More about extinction of opioid CPP below (Section 9.1.1)). Reinstatement of morphine CPP is prevented by downregulation of DRD1 MSN transmission (Hearing et al., 2016). CPP apparently depends upon DRD1s and DRD2s, as knocking them out or antagonizing them pharmacologically impairs morphine-induced CPP (Acquas et al., 1989; Fenu et al., 2006; Kobrin et al., 2017; Maldonado et al., 1997; Wang et al., 2015), whereas heroin self-administration is increased through the addition of a DRD1 agonist to heroin (Rowlett et al., 2007). Interestingly, after brain injury, accumbal plasticity is altered by inflammatory mechanisms. Mice demonstrated increased cocaine CPP compared to saline controls (Merkel et al., 2017) – a potential accumbal mechanism involved in neuroinflammatory responses. Studies such as these have not examined effects on morphine reward.
Molecular mechanisms for opioid reward involve epigenetic modifications of the genome. For example, heroin dose-dependently increased CPP, and Histone H3 phosphoacetylation was increased in the NAc of heroin group vs. controls (Sheng et al., 2011). Additionally, Sirtuins (SIRTs) are histone deacetylases, and influence brain function in opioid addiction. Chronic morphine administration producing CPP induces accumbal SIRT1 expression (Ferguson et al., 2013), while knockdown of SIRT1 in the NAc of floxed SIRT1 mice reduces drug reward. These behavioral effects of SIRT1 correspond with its ability to regulate trophic genes such as brain derived neurotrophic factor (BDNF).
7. Opioid abstinence and accumbal plasticity
Abstinence from chronic opioid exposure precipitates the experience of withdrawal (Kaplan et al., 2011a). Withdrawal from opioids is characterized by negative affective and physical symptoms, such as anxiety, dysphoria, vomiting, diarrhea, chills, muscle cramps and spasms, tremor, insomnia, as well as other subjective symptoms, including increased pain and stress sensitivity (Koob et al., 2014). Opioid withdrawal can be modeled in nonhuman animals. For example, morphine pellet-implanted mice, as compared to vehicle pellet-implanted mice, show significant increases in opioid withdrawal after administration of the opioid antagonist, naloxone. Somatic signs of jumping, wet-dog shakes, forepaw tremors, and diarrhea increase after opioid antagonist naloxone injection (Kaplan et al., 1994). It is believed that these aversive affective and physical states function as negative reinforcers, which motivate a return to addictive involvement, and have been shown to accompany drug-induced neuroadaptations to the Glu, DA, NE, and CRF systems, ostensibly emerging to neutralize the addictive effects of the drug (George and Koob, 2017; Koob and Moal, 2005). Such neuroadaptations generally are revealed through abstinence and include decreased baseline levels of extracellular DA at both pre- and postsynaptic levels, decreased availability of DA receptors, an inefficiency of the DA neurons, and an overall downregulation of the DA system (Koob, 2013; Koob and Le Moal, 2008; Melis et al., 2005). It has been hypothesized that addicted individuals are motivated to compensate for the negative experiences associated with this hypodopaminergic state by becoming re-involved with the substance or activity of addiction, to once again feel normal or to “get straight” through an attempt to return to his/her baseline hedonic setpoint (Koob et al., 2014). Withdrawal relief craving, therefore, can be understood to emerge from this hypodopaminergic state. There are other potential accumbal mechanisms related to opioid withdrawal that could relate to changes in synaptic plasticity. A reduction in spine density of the NAc shell, but not the core, is associated with the hypodopaminergic state that occurs during both spontaneous and naloxone-induced withdrawal (Spiga et al., 2005). Morphine withdrawal results in an enduring yet reversible reduction of spines’ density in shell MSNs, which can persist for up to 14 days, after which spine density returns to pretreatment levels (Diana et al., 2006). These structural and synaptic events likely correspond with the behavioral consequences of drug craving and loss of inhibitory control over intake (Diana et al., 2006).
The experience and behavioral symptoms of opioid withdrawal involve transcription factors and epigenetic mechanisms. For example, alterations in a histone methyltransferase (G9a) that catalyzes methylation of histone H3 at lysine 9 has been implicated in neural and behavioral plasticity. In a study conducted by Sun et al. (2012), mice in which G9a was overexpressed in the NAc were injected intraperitoneally with escalating doses of morphine. Two hours after the final morphine injection, the opioid antagonist naloxone was injected, upon which the mice with accumbal G9a overexpression (vs. control condition) exhibited an increase in withdrawal behaviors, including jumps, ptosis, tremors, diarrhea, and weight loss. Interestingly, in this same study, analgesic tolerance was tested using a hotplate paw lick test, in which repeated morphine injections (15 and 20 mg/kg s.c.) were administered for four days, and analgesia was measured 30 min after each drug dose. More rapid development of tolerance to morphine was exhibited by mice with overexpression of accumbal G9a compared to GFP controls (Sun et al., 2012).
Opioid withdrawal also involves small GTPases, which are intracellular targets of drugs of abuse that induce transcription and spine morphogenesis. For example, in a study conducted by Cahill et al. (2018), RhoA small GTPase was activated during morphine withdrawal-induced dendritic spine remodeling in the NAc. This group also observed that the RhoA network is engaged in NAc synaptic regions during protracted morphine withdrawal (two weeks), and that mice with an overexpression of accumbal RhoA GTPase exhibit reductions in the density of thin spines relative to controls (Cahill et al., 2018).
7.1. Models of abstinence in opioid use disorder
7.1.1. Opioid conditioned place aversion
An experimental measure of the negative affective experiences of withdrawal comes from performance on a variation of the CPP paradigm, known as conditioned place aversion (CPA). In a CPA procedure used by Hand et al. (1988), several days after morphine pellet implantation, rats were confined to the naloxone-paired compartment of a CPP box immediately following receipt of naloxone. The change of preference induced by the drug was reflected by the difference between the time spent in the naloxone-paired compartment after conditioning, minus the time spent in the same compartment before conditioning. Place aversion, or the affective component of drug withdrawal, is indicated by a negative score (Hand et al., 1988; Koob et al., 2014).
These methods have been utilized to determine whether either condition of withdrawal results in structural and/or synaptic plasticity within sites such as the VTA and locus correleus (Mazei-Robison and Nestler, 2012), as well as the NAc. Spiga et al. (2005) found reductions in spine density in both conditions, which was localized to MSN second-order dendrites in the NAc shell, but not the core. Diana et al. (2006) found similar reductions in NAc shell spine density, in addition to the fact that these alterations lasted up to 14 days following the most recent morphine administration, before reversing themselves. These findings were consistent with previous findings that long-term (24–25 days) intermittent withdrawal from repeated and intermittent administration of morphine results in reductions of both the branching and density of dendrites within the NAc shell (Robinson et al., 2002; Robinson and Kolb, 1999). After 21–28 days of repeated morphine administration, there was a reduction in total density of accumbal MSN spines, mostly due to the attenuation of long-thin spines (Graziane et al., 2016). In that study, co-administration of Glu-A2/3 peptide with morphine blocked these effects, thereby suggesting prevention of AMPAR internalization and synapse elimination after morphine withdrawal.
The structural changes to dendritic spines in the NAc accompanying withdrawal are associated with synaptic changes. For example, protracted withdrawal from heroin is associated with persistent reductions in spine head diameter, as well as the AMPAR:NMDAR ratio (Shen et al., 2011). Early and extended withdrawal from self-administered heroin, which precedes both LTP and LTD induced by drugs within the NAc (Shen and Kalivas, 2013), is associated with increased NMDA2B-containing receptors in the NAc (Shen et al., 2011). Dong et al. (2007) found that acute (12 h) and prolonged (four days) morphine withdrawal induced changes of NAc function by obstructing synaptic plasticity through endogenous LTP and LTD in the subicular-NAc pathway. Wu et al. (2012) showed that protracted morphine withdrawal (10 days) downregulated metabotropic GluR2/3 NMDARs, thus inducing potentiation in the glutamatergic synaptic strength through increased Glu release, resulting in the intrinsic excitability of MSNs within the NAc shell.
Other glutamatergic mechanisms in the accumbens are important in the negative, aversive effects of opioid withdrawal. AMPAR antagonist injection into the shell of the NAc of morphine-dependent rats blocked naloxone-induced CPA and reduced sensitivity to brain stimulation reward (Russell et al., 2016). Chronic morphine increased accumbal GluA1 levels while in morphine-dependent rats, naloxone treatment produced compensatory decreases in accumbal AMPA GluA1 levels. These results suggest that synaptic changes in accumbal GluA1 AMPARs are critical to the negative affective states of opioid withdrawal.
The input from the paraventricular nucleus of the thalamus (PVT) to the NAc appears to be a key pathway in the facilitation of the aversive states associated with opioid withdrawal (Zhu et al., 2016). For example, the administration of naloxone to morphine-dependent mice induced somatic signs and avoidance in the context of the withdrawal chamber of the CPA test, along with the expression of neural activity marker c-Fos in PVT-NAc projection neurons. Moreover, in this study, bilateral optogenetic silencing of the PVT-NAc pathway during naloxone-precipitated withdrawal blocked somatic signs of opioid dependence and CPA, and morphine-treated mice potentiated PVT-DRD2 MSN synapses, but not PVT-DRD1 MSN synapses. These findings suggest that novel treatments for OUD may involve the targeted stimulation of these pathways to induce plasticity.
8. Opioid craving and reinstatement of involvement
Unlike the majority of individuals who use opioids on a regular basis for medical reasons (Grant et al., 2016; Morissette et al., 2014), individuals with an OUD report being driven to use opioids by the phenomenon of craving, or rather, by ‘wants they do not want to want’ (Frankfurt, 1988). Importantly, there is no universally agreed upon definition of craving. For the purposes of this review, we understand craving to refer to an intense, urgent “abnormal desire” characterized by longing, yearning, and physiological need to become involved with a substance or activity (Anton, 1999; Sinha, 2013). Craving pertains to many behavioral elements of addiction, including drug-seeking, drug-taking/administration, and relapse/reinstatement. Indeed, craving functions as a unifying principle perpetuating the addiction cycle in a recursive manner between positive and negative affective experiences. As we have observed, distinct subtypes of craving tend to emerge within each stage (i.e., reward craving and withdrawal relief craving). Due to its subjective nature, measures of craving in human studies typically rely on self-report, while nonhuman animal studies tend to rely on objective behavioral measures, such as drug-seeking and drug-taking behaviors. Although craving is partly due to the impaired inhibitory function of specific components of the mesocorticolimbic system (including orbitofrontal frontal cortex, ventromedial frontal cortex, and anterior cingulate cortex) (Jentsch and Taylor, 1999; Volkow et al., 2011), we are presently concerned with the impairments in behavioral inhibition that are linked to alterations of the NAc MSN dendritic spines.
Cell-type specific Glu molecules play a role in relapse models. In one study, morphine CPP treatment, there was a 14-day abstinence period. Following this abstinence, a morphine prime was given and there was a reinstatement of CPP along with endocytosis of GluA2-containing AMPARs in DRD2-MSNs in the shell. This effect was blocked by an intra-NAC shell infusion of the Tat-GluA23Y peptide (Madayag et al., 2019). This study highlights the role of an accumbal GluA2 molecule in MSN that may play a role in relapse. Similar findings by Hearing et al. (2016) showed that, in transgenic mice, repeated morphine treatment, followed by abstinence and then a morphine prime, resulted in enhanced NAc shell MSN synaptic strength and AMPAR signaling. In this study, this reinstatement increases AMPAR:NMDAR electrophysiological ratios in DRD1 MSNs. In GluA2-lacking AMPAR in DRD1 MSNs, morphine reinstatement reduced electrophysiological measures during reinstatement.
An IEG called activity-regulated cytoskeleton-associated protein (Arc) is selectively targeted to synaptic sites where it can be translated. This enables Arc to link itself to synaptic activity to protein synthesis and synaptic plasticity. Morphine CPP increases Arc/Arg3.1 protein in the NAc shell. After an 8-day period of extinction from morphine CPP, intra-NAc core injection of Arc/Arg3.1 antisense oligodeoxynucleotide (AS) blocked morphine prime induced reinstatement of CPP (Lv et al., 2011). Finally, trophic factors appear to play a role in morphine relapse mechanisms. In one example by Meng et al. (2013), mice exposed to morphine CPP were given a two-week extinction period and then a priming dose of morphine with CPP measurement. Levels of BDNF mRNA splice variants increased during CPP and then decreased after extinction training. The levels continued to decrease during reinstatement induced by a morphine priming injection (10 mg/kg i.n.). Thus, changes in accumbal BDNF plasticity in the NAc plays a role in opioid reinstatement.
9. Recovery from opioid use disorder and accumbal plasticity
Despite the fact that many people do recover from OUD, recovery is the least understood stage of addiction. The construct of recovery has been variously defined (Kaskutas et al., 2015; Kelly and Hoeppner, 2015; Knopf, 2011; Laudet, 2007; The Betty Ford Institute Consensus Panel, 2007; White, 2007), but, in general, it is described in behavioral terms, as a reduction or elimination of the behaviors associated with addictive involvement, which are represented by a continuum of changes ranging from total abstinence, to moderation, to intervention strategies utilizing pharmacological adjuncts (e.g., naltrexone, methadone, buprenorphine) (White and Kurtz, 2006) and deep brain stimulation (Kuhn et al., 2014). It is likely that the behavioral changes associated with recovery are accompanied by plasticity changes of the dendritic spines of the NAc.
As discussed above, early and protracted abstinence from opioids, which generally characterize the withdrawal stage of addiction, have been shown to induce plasticity changes within the NAc. It is unknown, however, whether long-term abstinence (i.e., greater than five years) from opioids is associated with its own distinctive plasticity changes. It is also unknown whether specific treatment modalities, which have been proven effective in ameliorating OUD symptoms, are associated with unique accumbal plasticity changes. For example, it remains to be seen whether drugs such as methadone and buprenorphine, frequently prescribed to treat OUD and known to attenuate craving through similar pharmacological mechanisms as abused opioids, produce similar or unique plasticity changes to morphine and heroin. Comparable questions pertain to the long-term therapeutic use of an opioid antagonist, such as naltrexone, as well as involvement in mutual-help organizations (e.g., Narcotics Anonymous), which ostensibly represents a human analogue to the environmental enrichment model that has been correlated with both reduction in drug self-administration in addition to alterations in DAergic signaling and receptor expression in nonhuman animal studies (El Rawas et al., 2009; Morgan et al., 2002; Tomek and Olive, 2018).
9.1. Models of opioid use disorder recovery
9.1.1. Extinction of opioid conditioned place preference
One aspect of treatment/recovery that has revealed changes in accumbal plasticity has been modeled through the extinction of opioid-induced CPP. In nonhuman animal models, when the conditioned preference is attenuated through repeated parings of the drug-associated cues or contexts and the absence of the drug, “extinction” is said to have occurred (Kobrin et al., 2017; Tzschentke, 2007). Extinction training is intended to reduce associations that were learned previously, and it functions as an animal model for cue exposure therapy, in which drug craving is attenuated in an addicted individual by means of exposure to drug-related cues and contexts in the absence of the previously conditioned drug reward (Kantak and Dhonnchadha, 2011; Kobrin et al., 2017; Vollstadt-Klein et al., 2011; Wolf, 2016).
Extinction learning of opioid CPP is dependent on NAc MSN morphology. For example, morphine CPP extinction is associated with a decrease in NAc core dendritic complexity (Kobrin et al., 2015; Leite-Morris et al., 2014). However, when DRD1 were activated, morphine-induced extinction was not only attenuated, but dendritic complexity was increased within the NAc core, but not the shell, which may correspond to an increase in accumbal synaptic inputs (Kobrin et al., 2017). These findings suggest that reward-associated behaviors are maintained by dopaminergic signaling, and extinction of these behaviors may result from a decline in accumbal dopaminergic signaling, along with changes in other pathways (Kobrin et al., 2017).
Martínez-Rivera et al. (2019) examined the effects of extinction of morphine CPP on gene expression associated with NAc synaptic plasticity. BDNF mRNA was increased in the NAc of rats that were able to extinguish their preference for the morphine associated context. A subgroup that were not able to extinguish that preference showed induction of the accumbal cytokine, Tumor Necrosis Factor alpha, the transcription factor, cAMP responsive element modulator, and the cell cycle protein, Ras homolog. This suggests differential accumbal plasticity in animals that could extinguish their preference for morphine and those that could not.
Lastly, buprenorphine is a partial mu-opioid agonist that is a mainstay of treatment in OUD in humans. In one animal model study, buprenorphine reduced the heroin-induced rise in NAc DA as measured by in vivo microdialysis. In rats taught to self-administer heroin, buprenorphine treatment reduced heroin seeking in the presence of conditioned drug cues during extinction. It also reduced seeking in priming-induced reinstatement induced by heroin (Sorge and Stewart, 2006). Future treatments may consist of drugs that act at many of the neuroplastic systems discussed in this review.
10. Conclusions
Opioids induce key molecular, cellular, and structural changes within the mesocorticolimbic DA and glutamatergic systems that converge in the NAc and contribute to an individual’s vulnerability to OUD. The combination of morphine treatment paradigm, dosage, and associated learning produces unique molecular changes and it leads to altered epigenetics, gene expression, plasticity, and circuit remodeling with corresponding changes in behavior. We have examined evidence that opioids contribute to structural and functional plasticity of the dendritic spines within the NAc, a region of the brain that has been shown to be essential for processing rewards and translating motivations into goal-directed action. The addiction-related behavioral adaptations that accompany the opioid-induced neuroplastic changes in the NAc, which are evident in preclinical studies, suggest a likely pathway through which OUD develops among humans.
We have provided an overview of the opioid-induced signaling and circuit changes within the context of addiction-related behaviors (i.e., involvement and abstinence) and relevant experimental paradigm (i.e., self-administration, sensitization, CPP, CPA, withdrawal, etc.). We have provided numerous examples of studies demonstrating that the structural and synaptic plasticity of dendritic spines within the NAc depends on a variety of molecular mechanisms involving specific receptors, effectors, G proteins, transcription factors, epigenetic processes, and cytoskeletal proteins. Many studies have shown that opioids regulate accumbal epigenetic processes and transcriptional activity for specific plasticity-related gene expression resulting in dendritic changes. There are well-established links between addiction-related behavioral profiles and abnormal spine morphology, neuroplastic proteins contributing to spine morphology, and electrophysiological alterations in neurons and circuits. Functional changes in accumbal neurons are dependent on LTP or LTD, structural rearrangements of actin filaments in dendritic spines and NMDAR activation (Cingolani and Goda, 2008).
Given the growing prevalence of OUD within society, along with its attendant economic implications, there is tremendous urgency to provide the safest and most effective treatment possible. Obviously, more research is needed to that end. Our objective in this article has been to clear the way a bit by describing the findings that exemplify what is currently known about the neurobiological underpinnings of each phase of opioid addiction. Our motivation for this approach has been to synthesize a vast and complex body of evidence in such a way as to provide clear direction for future research in this field, in addition to making translational and clinically meaningful insights as practical as possible.