Abstract
Methamphetamine (METH) is an illicit psychostimulant that is abused throughout the world. METH addiction is also a major public health concern and the abuse of large doses of the drug is often associated with serious neuropsychiatric consequences that may include agitation, anxiety, hallucinations, paranoia, and psychosis. Some human methamphetamine users can also suffer from attention, memory, and executive deficits. METH-associated neurological and psychiatric complications might be related, in part, to METH-induced neurotoxic effects. Those include altered dopaminergic and serotonergic functions, neuronal apoptosis, astrocytosis, and microgliosis. Here we have endeavored to discuss some of the main effects of the drug and have presented the evidence supporting certain of the molecular and cellular bases of METH neurotoxicity. The accumulated evidence suggests the involvement of transcription factors, activation of dealth pathways that emanate from mitochondria and endoplasmic reticulum (ER), and a role for neuroinflammatory mechanisms. Understanding the molecular processes involved in METH induced neurotoxicity should help in developing better therapeutic approaches that might also serve to attenuate or block the biological consequences of use of large doses of the drug by some humans who meet criteria for METH use disorder.
1. General introduction
Methamphetamine (METH) is a psychostimulant that is abused worldwide (UNODC, 2018; Yang et al., 2018). METH was first synthesized from ephedrine by Japanese chemist, Nagayoshi Nagai in 1893. In 1919, another Japanese chemist, Akira Ogata streamlined the process and produced the first crystallized form of drug (Nagai and Kamiyama, 1988; Buxton and Dove, 2008; Panenka et al., 2013). The use of METH, which is also a schedule II drug, has been restricted by USA law since 1971. METH is used as a second line of treatment for attention deficit hyperactivity disorder (ADHD), severe obesity, and narcolepsy(Moszczynska and Callan, 2017). Its repeated use, under uncontrolled conditions, can lead to the user meeting diagnostic criteria for METH use disorder which is characterized by compulsive use in the presence of adverse consequences and craving for the drug (DSM–5, 2013). Some of the adverse consequences include neurological and psychiatric complications, cardiovascular problems, ulmonary arterial hypertension, periodontal (gum) disease, and renal failure (Ho et al., 2009; Schep et al., 2010; Moratalla et al., 2017; Yang et al., 2018). Specific METH-induced neurological and psychiatric effects include cerebral stroke, seizures, schizophrenia, and psychotic illness (Cadet and Gold, 2017; Hsieh et al., 2014; Yang et al., 2018; Lappin and Sara, 2019; Wearne and Cornish, 2018).
In addition to the clinical signs and symptoms associated with the large doses of METH, several lines of evidence have documented its toxic effects on dopamine (DA) and serotonin (5-HT) systems (Cadet and Krasnova, 2009). Large METH doses can cause neuronal apoptosis and glial activation in the brain (Panenka et al., 2013; Moratalla et al., 2017; Sekine et al., 2008; Yang et al., 2018). Moreover, acute and chronic injection of the drug have induced a diversity of toxic responses in animal models (Cadet et al., 2003, Cadet et al., 2005, Cadet et al., 2007; Cadet and Bisagno, 2015). Despite knowing the potential toxic effects of the drug, little effort has been spent to develop pharmacological approaches in order to counter these effects in human users. Studies focusing biological mechanisms of METH induced neurotoxicity should provide the knowledge necessary to approach these problems more rationally (Ashok et al., 2017; Moszczynska and Callan, 2017; Yang et al., 2018; Xie et al., 2018).
2. Epidemiology of METH use
METH is a member of the amphetamine-type stimulants (ATSs) that include amphetamine, methylene dioxy methamphetamine (MDMA), and other designer amphetamine (Chomchai and Chomchai, 2015; Yang et al., 2018). It has been reported that approximately 27 million individuals use ATSs in 2019, a number that corresponds to 0.5 per cent of the world adult population (UNODC, 2020). North America with 2.3 per cent, Australia and New Zealand with 1.3 per cent, and Asia with 0.5 per cent of their populations have the highest prevalence of METH use between ages 15 and 64 (UNODC, 2020). METH is indeed the second most used illicit drug after cannabis (Stoneberg et al., 2018). METH manufacturing contributes to 95% of illigally synthesized ATS, with the quantities of the drug having been seized between 2009 and 2017 increasing by sevenfold (UNODC, 2020). Seizures of illegal METH remain highly concentrated in the United States, Thailand, and Mexico, accounting for 80% of total global seizures (UNODC, 2020). The compounds utilized to synthesize METH, ephedrine and pseudoephedrine, are used in Asia, Oceania, Africa and in some European regions whereas phenyl-2-propanone (P-2-P), a pseudoephedrine precursor, is mostly used in North America and Western Europe (EU Drug Markets Report 2019: E/INCB/2019/1, 2019; UNODC, 2020).
In the USA, nearly 1.6 million individuals were reported to use METH in 2016, with an average age of 23.3 years old according to the National Survey on Drug Use and Health (2017). In 2017, METH-related overdose deaths in United states increased by 7.5 times compared to 2007. These cases occurred mostly in Washington, Colorado, Texas, Florida, and Georgia (National Survey on Drug Use and Health, 2017).
3. METH use and its clinical neuropsychiatric presentations
METH use is associated with several health complications secondary to the negative impact of the drug on the central nervous system (CNS). These include cognitive and psychomotor impairments users of large doses of the drug (Panenka et al., 2013; Yang et al., 2018; Paulus and Stewart, 2020). Human METH users can suffer drug-induced agitation, anxiety, paranoia, and psychosis (Paulus and Stewart, 2020; Zhao et al., 2020). METH users have presented to emergency rooms with strokes, seizures, renal and liver failure, cardiac arrythmias, extreme hyperthermia, or in comatose states (Perez Jr et al., 1999; Turnipseed et al., 2003; McGee et al., 2004; Ho et al., 2009; Schep et al., 2010; Jones and Rayner, 2015). A meta-analysis has reported that METH users can suffer from neuropsychological impairments consisting of dysfunctions of decision making, information processing speed, language, and visuoconstructional abilities (Scott et al., 2007).
Importantly, some recent reports have documented a higher prevalence of Parkinosism in METH users (Callaghan et al., 2010, Callaghan et al., 2012; McNeely et al., 2012; Panenka et al., 2013; Curtin et al., 2015; Todd et al., 2016). For example, Callaghan et al. (2012) reported that METH abusers have a 75% higher risk of developing Parkinsonism than non-METH using individuals. Retrospective case-controlled studies have also found that prolonged use of METH was also associated with an increased risk for developping Parkinson’s disease (PD) (Garwood et al., 2006; Curtin et al., 2015). Neurodegenerative changes consisting of loss of dopamine transporters (DAT), serotonin transporters (5-HTT), and decreased levels of dopamine (DA) and its metabolites have been detected in the brains of human METH users (Wilson et al., 1996; Worsley et al., 2000; Volkow et al., 2001; Sekine et al., 2003, Sekine et al., 2006).
METH users also exhibit prominent gray matter reduction in the cortical (Berman et al., 2008) and hippocampal (Thompson et al., 2004; Hall et al., 2015) brain regions. Other investigators have reported higher striatal volume was observed in METH abusers (Thompson et al., 2004). Moreover, Tobias et al. (2010) reported that METH users exhibited decreased fractional anisotropy in the prefrontal white matter, the midline genu of the corpus callosum, and in the midcaudal superior corona radiata bilaterally. It is possible that some of the neuropathological changes observed in the brains of METH users might be secondary to the activation of microglial cells observed in the brains of some of these patients (Sekine et al., 2008).
4. Animal models of METH neurotoxicity
Starting from the 1970’s, various studies have been published to show that both acute and chronic injections of METH can cause damage to monoaminergic terminals and neuronal apoptosis (Seiden et al., 1976; Ando et al., 1985; Woolverton et al., 1989; Fukumura et al., 1998; Villemagne et al., 1998; Harvey et al., 2000a, Harvey et al., 2000b; Ladenheim et al., 2000; Armstrong and Noguchi, 2004; Jayanthi et al., 2001, Jayanthi et al., 2005; Truong et al., 2005; Deng et al., 2001, Deng et al., 2007; Melega et al., 1997, Melega et al., 2008; Ares-Santos et al., 2013; Schweppe et al., 2020).
4.1. Studies in rodents
Injections of large METH doses induce degeneration of monoaminergic terminals in rodents (Fukumura et al., 1998; Ladenheim et al., 2000; Armstrong and Noguchi, 2004; Jayanthi et al., 2001, Jayanthi et al., 2005; Truong et al., 2005; Deng et al., 2001, Deng et al., 2007; Ares-Santos et al., 2013). These are characterized by longterm decreases in vesicular DA uptakeand vesicular monoamine transporters (VMAT2) (Guilarte et al., 2003; Truong et al., 2005), striatal DA transporters (Fukumura et al., 1998; Truong et al., 2005; Krasnova et al., 2011), levels of tyrosine hydroxylase (TH) protein and activity (Hotchkiss and Gibb, 1980; Fukumura et al., 1998; Cappon et al., 2000; Krasnova et al., 2011). Large doses of METH also negatively impact serotonerigc systems in the dorsal striatum where striatal serotonin (5-HT) levels (Fukumura et al., 1998: Armstrong and Noguchi, 2004) and tryptophan hydroxylase (TPH) activity (Bakhit et al., 1981; Bakhit and Gibb, 1981) are reduced after injections of the drug. METH-induced abnormalities in 5-HT have also been reported in the nucleus accumbens (Nac), cortex, hippocampus, and hypothalamus (Hotchkiss and Gibb, 1980; Bakhit et al., 1981; Bakhit and Gibb, 1981; Baldwin et al., 1993; Armstrong and Noguchi, 2004). A very recent paper by Schweppe et al. (2020) reported that rats challenged with toxic doses of METH showed reduction in striatal and hippocampal DA, 5-HT, brain derived neurotrophic factor (BDNF), and TrkB even as long as 75 days after the drug injections. METH neurotoxicity is also associated with astrocytic and microglial activation (Fukumura et al., 1998; Guilarte et al., 2003).
Consistent with data obtained from rats, mice also suffer from METH-induced decreased levels of DA and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid(HVA), as well as reduced levels of VMAT2, DAT and TH activity in various brain regions including the dorsal striatum, cortex, hippocampus and the olfactory bulb (Ladenheim et al., 2000; Achat-Mendes et al., 2005; Deng et al., 2007; Fantegrossi et al., 2008; Granado et al., 2010, Granado et al., 2011a, Granado et al., 2011b; Ares-Santos et al., 2012; Ares-Santos et al., 2013).
The neurotoxic effects of METH have also been assessed in rodent model of METH self-administration (SA). Specifically, rats given long access to METH SA in order to mimic patterns of METH use in humans (Perez Jr et al., 1999; Darke et al., 2008) exhibited persistent decreases in DA, DAT and TH but increased glial fibrillary acidic protein (GFAP) expression in the cortex and dorsal striatum and cortex (Krasnova et al., 2010). Similar to the report by Krasnova et al. (2010), McFadden et al. (2012) also observed persistent deficits in dopaminergic neuronal function consistent of decreased striatal DAT uptake, DAT concentrations, and increased GFAP using a SA paradigm of 8 h/day for 7 days (0.06 mg/infusion). Together, these two SA studies provide further evidence for the toxic effects of this drug.
4.2. Studies in primates
Rhesus monkeys injected with METH showed 70% loss of DA levels in the caudate, 33% loss of NE in the midbrain and 55% loss of NE in the frontal cortex (Seiden et al., 1976). There were signficant decreases in DA and 5-HT level in various brain regions of nonhuman primates even 4 years after the injections of METH (Woolverton et al., 1989). Other groups of investigators have confirmed the effects of METH on the nonhuman primate brain. For example, Ando et al. (1985) reported a 32% loss of DA levels in the caudate nucleus and 71% loss of 5-HT levels in the frontal cortex after METH. In addition, Harvey et al., 2000a, Harvey et al., 2000b have provided evidence that METH injections can cause decreased levels TH, DAT, and VMAT2 in the nigrostriatal dopaminergic system. In vivo positron emission tomography (PET) studies in vervet monkeys also reported METH-induced reduced DA synthesis (Melega et al., 1997, Melega et al., 2008). Furthermore, administration of METH to baboons exhibited reductions in striatal DAT which is associated with decreased level of DA (Villemagne et al., 1998).
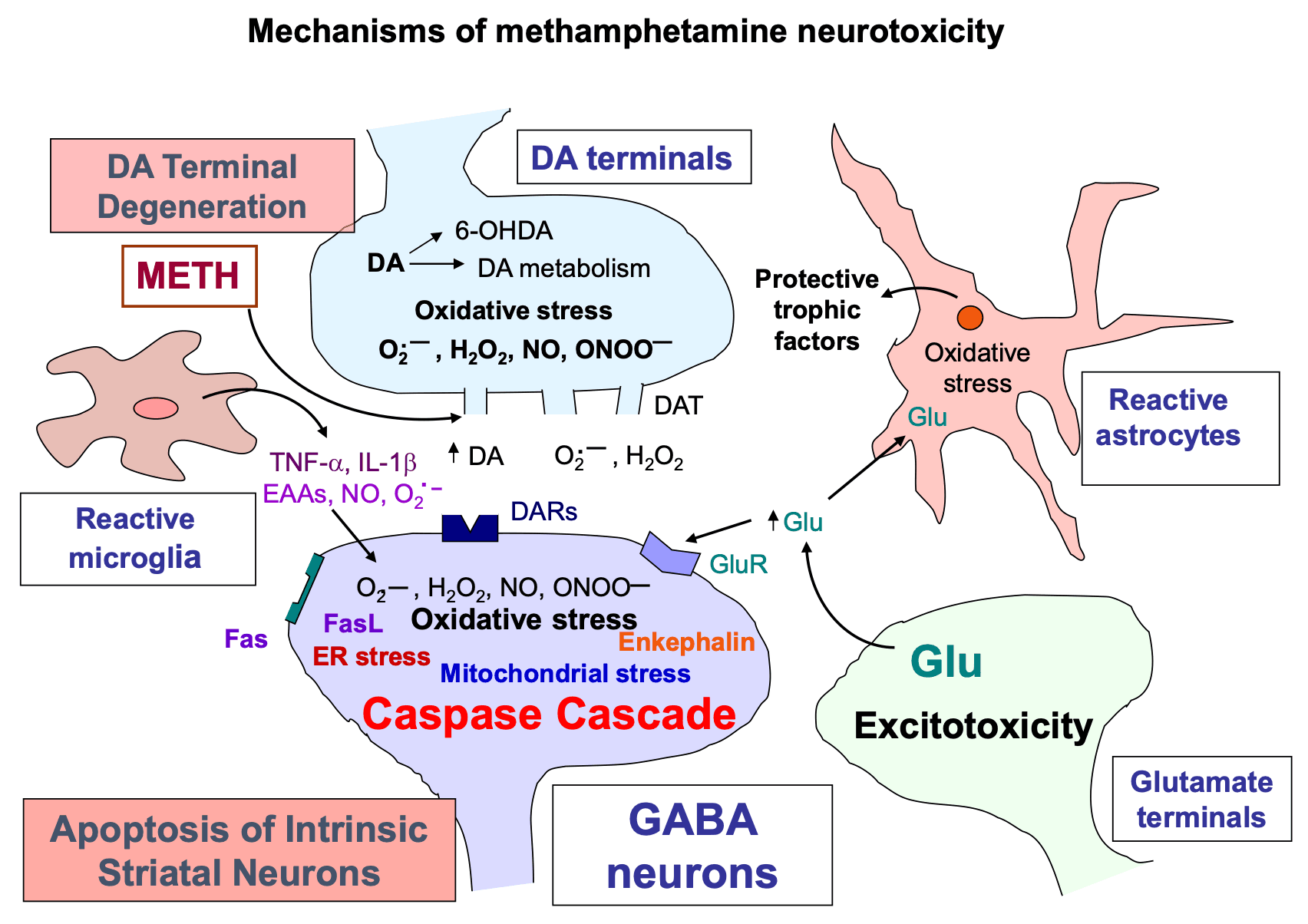
5. METH neurotoxicity, reactive oxygen species, and neuroinflammation
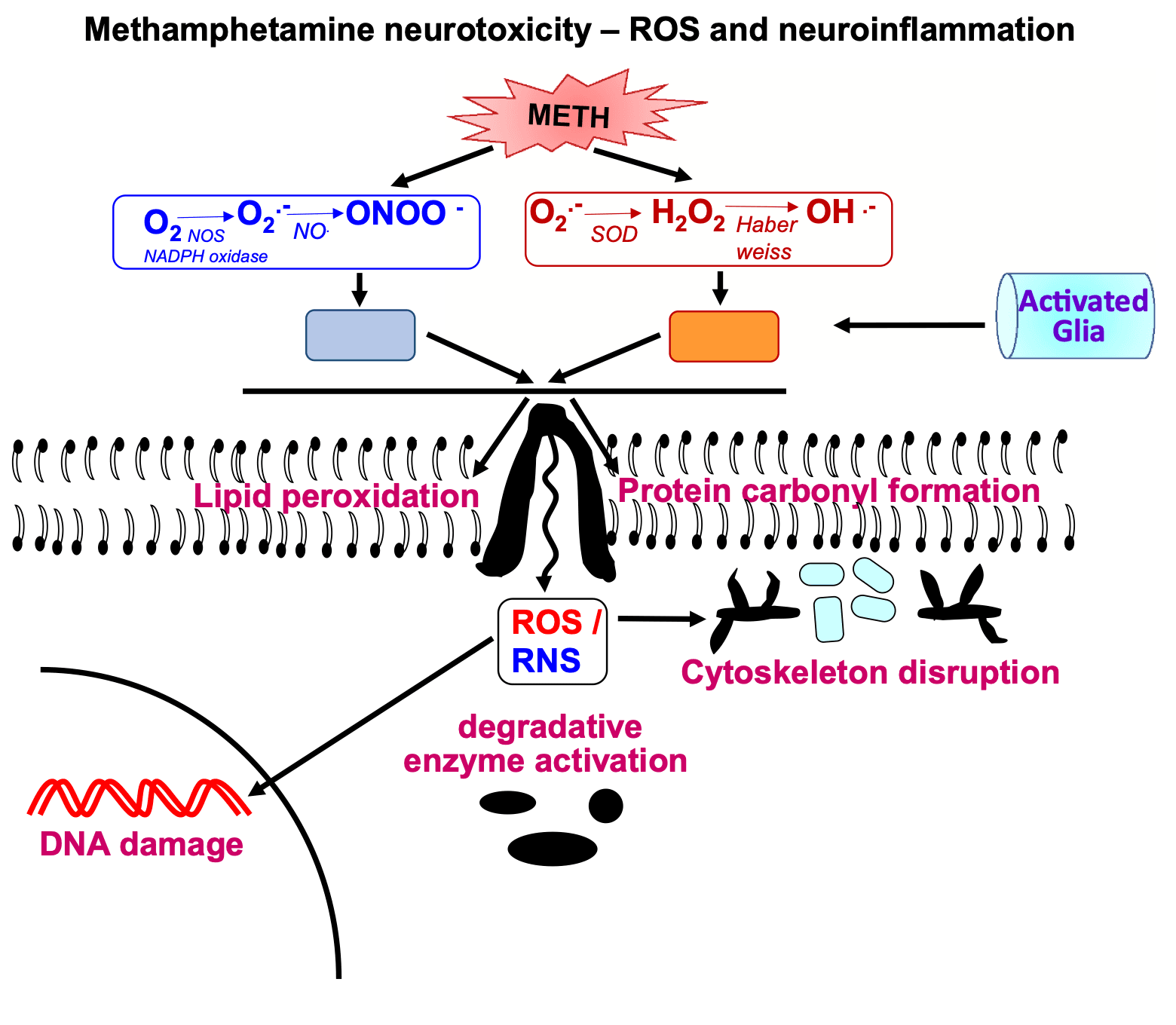
The pathways involved in causing METH neurotoxicity are varied and complex. They include the formation of reactive oxygen species including hydrogen peroxide, superoxide radicals, and hydroxyl radicals. The levels of some of these appear to be increased consequent to microglial cell activation and associated changes in proinflammatory factors in brain regions of interest.
5.1. Production of reactive oxygen species (ROS)
Oxidative stress plays an integral role in METH neurotoxicity. This occurs because METH administration leads to release of DA from vesicular pools followed by DA accumulation within monoaminergic terminals and DA release via DAT into the synaptic cleft (Chu et al., 2008; Hedges et al., 2018). Increased DA levels lead to DA auto-oxidation in intraneuronal and extracellular spaces, quinone production, superoxide radicals, hydrogen peroxide, and hydroxyl radicals (Graham, 1978; Cadet and Brannock, 1998; Yamamoto and Zhu, 1998; LaVoie and Hastings, 1999). The role of superoxide radicals in METH neurotoxicity was documented in a series of studies that showed that transgenic mice that over-express superoxide dismutase, the enzyme that breaks down superoxide radicals (Lewandowski et al., 2019), were protected against injections of large doses of METH (Cadet et al., 1994; Hirata et al., 1996; Jayanthi et al., 1998). Reactive nitrogen species also participate in generating METH neurotoxicity. This occurs through the production of nitric oxidesecondary to METH-induced increases in nitric oxide synthase (NOS) activity (Imam et al., 2001). METH-induced ROS and RNS lead to lipid peroxidation and protein carbonyl formation in various brain regions and secondary damage to neuronal cell membrane (Jayanthi et al., 1998; Yamamoto and Zhu, 1998; Gluck et al., 2001). METH-induced impairment of blood-brain barrier (BBB) permeability may also occur via its pro-oxidant effects via activation of NADPH oxidase (Ramirez et al., 2009; Park et al., 2012; Jumnongprakhon et al., 2016).
Moreover, human chronic METH users have been reported with increased levels of oxidative stress markers (4-hydroxynonenal and malondialdehyde) (Fitzmaurice et al., 2006) along with decreased activity of phospholipid metabolic enzymes (Ross et al., 2002) and antioxidant systems in their brain (Mirecki et al., 2004).
5.2. Participation of microglial cells in METH-induced neurotoxic events
METH exposure causes both microglial and astrocyte activation in the brain (Sekine et al., 2008; Krasnova et al., 2010; McFadden et al., 2012). This is associated with increased production and secretion of pro-inflammatory cytokines that can cause neurodegeneration(Xu et al., 2017; Tahmasebinia and Pourgholaminejad, 2017; Temmingh et al., 2020; Fukumura et al., 1998). Release of pro-inflammatory cytokines can then activate apoptotic signaling cascades in several model systems (Allagnat et al., 2012; Park et al., 2017; Butovsky and Weiner, 2018).
METH administration increases the expression of glial fibrillary acidic protein (GFAP) in various brain regions (Fukumura et al., 1998; Guilarte et al., 2003; Krasnova et al., 2010; McFadden et al., 2012). METH also causes microglial activation in various brain regions (Gonçalves et al., 2017; Gou et al., 2020; Sekine et al., 2008; Thomas et al., 2004, Thomas et al., 2009). A role for microglial in the appearance of METH neurotoxicity is supported by reports that drugs such as MK-801 and dextromethorphan that block microglial activation can protect against the toxic effects of the drug (Asanuma et al., 2003; Thomas and Kuhn, 2005). The sigma-1 receptor may also participate in mediating METH neurotoxicity via their effects on glial activation. Specifically, sigma receptor-1 antagonists such as BD1047 and SN79 that block glial activation and expression of cytokines have been shown to protect against METH-induced neurotoxicity (Kaushal et al., 2013; Robson et al., 2013, Robson et al., 2014; Zhang et al., 2015).
6. METH neurotoxicity and cell death mechanisms
Biochemical studies using a diversity of animal models have provided evidence for the participation of multiple mechanisms in METH- induced neuronal cell death. These include pathways regulated by mitochondrial and endoplasmic reticulum (ER) proteins, and involvement of some transcription factors.
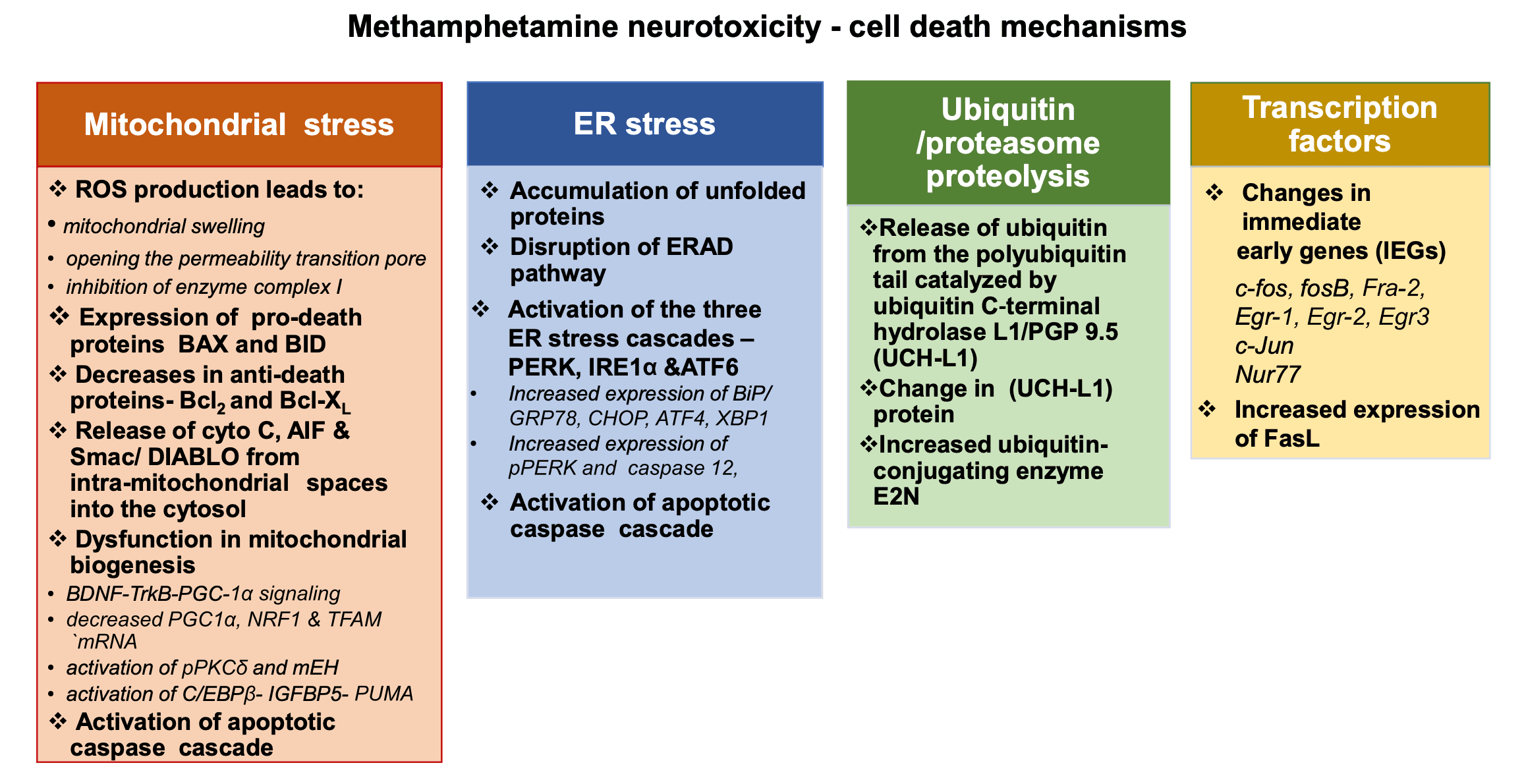
6.1. Mitochondrial stress and METH neurotoxicity
Mitochondrial dysfunction plays a critical role in METH-induced neurotoxicity (Cadet et al., 2005, Cadet et al., 2007). Auto-oxidation of excessive cytosolic and extracellular DA produces DA quinone and other reactive oxygen species (ROS) (Cadet, 1988; Cadet and Brannock, 1998; Cadet and Lohr, 1987). Similar mechanisms appear to be involved in the clinical manifestations and basic neuropathology of other neurological and psychiatric disorders including schizophrenia and Parkinson’s disease (Cadet and Lohr, 1987; Evans, 1993; Perfeito et al., 2012). DA oxidation products can cause mitochondrial dysfunctions which include mitochondrial swelling, opening the permeability transition pore, and inhibition of enzyme complex I (Berman and Hastings, 1999; Jana et al., 2011; Khan et al., 2005). Evidence for a role for mitochondrial dysfunctions in METH neurotoxicity was provided in a series of studies that showed that METH injections can cause increased expression of pro-death proteins, BAX and BID, concurrent with decreases in anti-apoptotic proteins, Bcl-2 and Bcl-XL in the brains of rodents (Jayanthi et al., 2001, Jayanthi et al., 2005; Deng et al., 2002). These changes are associated with release of cytochrome c, apoptosis inducing factor (AIF) and Smac/DIABLO from intra-mitochondrial spaces into the cytosol followed by induction of neuronal apoptosis (Jayanthi et al., 2001; Jayanthi et al., 2004; Deng et al., 2002). Treatment with the antioxidant, melatonin, was able to attenuate these degenerative effects of METH (Wisessmith et al., 2009). A role for superoxide radicals in METH-induced cell death is also supported by the demonstration that METH-induced pathological changes were suppressed in copper-zinc superoxide dismutase transgenic mice (Deng and Cadet, 2000).
Of related interest, Brown et al. (2005) have reported that administration of high doses of METH can inhibit the enzymatic activity of mitochondrial complexes in the dorsal striatumvia glutamate receptor- and peroxynitrite-mediated mechanisms. Sepehr et al. (2020) has also reported that METH can cause dysfunctions in the mitochondrial respiratory chain via a BDNF-TrkB-PGC-1α signaling pathway. This cascade is initiated when BDNF binds to its receptor TrkB and activates CREB signaling followed by increased expression of proliferator-activated gamma receptor coactivator 1-alpha (PGC-1α). PGC-1α, which is a key regulator of mitochondrial biogenesis (Fanibunda et al., 2019) and of the mitochondrial uncoupling protein-2 (UCP-2) (Sepehr et al., 2020). Additionally, several in vivo and in vitro studies have documented disturbances in mitochondrial biogenesis consequent to METH injections; those include decreased mRNA expression of mitochondrial biogenesis-involved factors, PGC1α, NRF1 and TFAM (Beirami et al., 2018; Valian et al., 2017, Valian et al., 2019; Seyedhosseini Tamijani et al., 2019). METH-induced mitochondrial dysfunctions might also occur via activation of protein kinase C-delta (PKCδ) (Dang et al., 2016, Dang et al., 2018; Nam et al., 2015; Nguyen et al., 2015; Shin et al., 2014, Shin et al., 2019) and its phosphorylation (Dang et al., 2018). Interactions between phosphorylated PKCδ and microsomal epoxide hydrolase (mEH) and between cleaved-PKCδ and mEH appear to also be involved in METH-induced cell death (Shin et al., 2019). It is also possible that METH-induced mitochondrial apoptotic signaling pathway might involve activation of the CCAAT-enhancer binding protein (C/EBPβ) (Qiao et al., 2014; Chen et al., 2016; Xu et al., 2018). For example, C/EBPβ can up-regulate the expression levels of insulin-like growth factor-binding protein 5 (IGFBP5) and p53-up-regulated modulator of apoptosis (PUMA) (Qiao et al., 2014; Chen et al., 2016; Xu et al., 2018) that eventually leads to downstream activation caspasecascade.
6.2. ER stress and METH neurotoxicity
The accumulated evidence suggests that METH-induced cell death can also occur via the activation of the endoplasmic reticulum (ER) stress (Krasnova and Cadet, 2009; Yu et al., 2015; Yang et al., 2018). ER stress is mediated by three pathways initiated by protein kinaseRNA-like endoplasmic reticulum kinase (PERK), inositol-requiring transmembrane kinase/endonuclease 1 (IRE1), and activating transcription factor (ATF) 6 (Khanna et al., 2021; Shacham et al., 2021; Siwecka et al., 2021; van Anken et al., 2021). ER stress occurs consequent to accumulation of unfolded proteins within the ER lumen followed by disruption of the ER-associated protein degradation (ERAD) pathway, altered ER homeostasis, and neuronal apoptosis (Kim et al., 2008; Hacker, 2014). Similar occurrences have been observed in the case of METH-induced neuronal death. Specifically, METH injections are accompanied by increased expression of several ER stress genes, including those that encode the 78-kDa glucose-regulated protein (GRP-78)/BiP, CCAAT/ enhancer-binding protein homologous protein (CHOP), and ATF4 (Jayanthi et al., 2005; Jayanthi et al., 2009; Hayashi et al., 2010; Beauvais et al., 2011; Takeichi et al., 2012; Cai et al., 2016; Wen et al., 2019; Chen et al., 2021). ER stress is accompanied by activation of the upstream ER-specific caspase, caspase-12 (Nakagawa et al., 2000; Szegezdi et al., 2003) followed by cleavage of the executioner caspase, caspase-3 (Jayanthi et al., 2004). Recent in vitro studiesby Wongprayoon and Govitrapong (2017) have also documented the involvement of the ER pathway in METH-induced death of SH-SY5Y neuronal cells treated with toxic doses of METH. The death mechanism includes increased CHOP expression, spliced X-box binding protein 1 (XBP1), caspase-12, and caspase-3 (Wongprayoon and Govitrapong, 2017). It is important to note that METH-mediated ER stress has been shown to be dependent on the activation of the DA D1 receptor in the rat brain (Jayanthi et al., 2009; Cadet et al., 2010; Beauvais et al., 2011), thus suggesting the possibility of using similar agents to counteract the toxic effects of the drug in humans. Xiao et al. (2018) have also reported that toxic doses of METH can significantly up-regulate the expression of phosphorylated PERK and caspase-12 and these effects can be suppressed by silencing of cyclin-dependent kinase (CDK) 5, a kinase that specifically phosphorylates Tau protein (Hashiguchi et al., 2002). In addition, Liu et al. (2020) documented METH induced time and dose-dependent activation of the three ER stress cascades, PERK, IRE1α and ATF6 signaling pathways, in hippocampal neuronal cells (HT-22). METH-induced disruptions of ER functions are accompanied by altered ER calcium homeostasis (Chen et al., 2019). They found that secreted ER calcium-monitoring proteins (SERCaMPs), a marker of ER stress that is triggered by depletion of ER calcium (Henderson et al., 2014) was significantly increased by METH (Chen et al., 2019).
6.3. METH and the ubiquitin/proteasome proteolytic pathway
METH neurotoxicity appears to also involve dysfunctions of the ubiquitin/proteasome system (UPS), a system that degrades intracellular proteins is involved in regulation of a broad array of cellular processes that include regulation of transcription factors and intracellular quality control (Glickman and Ciechanover, 2002; Ciechanover, 2013; Tai and Schuman, 2008). Maintenance of UPS function is by release of ubiquitin from the polyubiquitin tail and is catalyzed by ubiquitin C-terminal hydrolase L1/PGP 9.5 (UCH-L1) (Glickman and Ciechanover, 2002). In the nervous system, this system is important in the modulation of synaptic plasticity (Tai and Schuman, 2008) and in controlling mechanisms involved in neurodegenerative processes (Schmidt et al., 2021). These facts are consistent with the demonstration that toxic doses of METH alter (UCH-L1) protein levels (Liao et al., 2005) accompanied by incomplete degradation of target proteins and accumulation of prion protein aggregates in DA-containing cells (Ferrucci et al., 2017). A proteomic analysis has also revealed that METH injections caused increased ubiquitin-conjugating enzyme E2N in various brain regions of rats (Li et al., 2008). Involvement of this system in METH neurotoxicity needs to be investigated further.
6.4. Transcription factors and their involvement in METH neurotoxicity
Injections of toxic of METH have been shown to alter the expression of several transcription factors including immediate-early genes (IEGs) in various brain regions (Bisagno and Cadet, 2019; Cadet et al., 2002, Cadet et al., 2010) where METH-induced terminal degeneration and/or neuronal cell death have been observed (Deng et al., 2001, Deng et al., 2002, Deng et al., 2007; Jayanthi et al., 2004; Jayanthi et al., 2005). Some of these transcription factors include c-fos, fosB, Fra-2, Egr-1, Egr-2, and Egr3 (Hirata et al., 1998; Cadet et al., 2001; Thiriet et al., 2001; Jayanthi et al., 2005; Beauvais et al., 2010; Cadet et al., 2010; McCoy et al., 2011; Martin et al., 2012). A role for c-fos in the METH-induced cell death was provided by Deng et al. (1999) who reported that METH-neurotoxicity was significantly exacerbated in heterozygous and homozygous c-fos knock-out mice, with the homozygous showing greater loss of striatal dopaminergic markers. The authors also showed c-fos knock-out mice exhibited more DNA fragmentation in nondopaminergic cells in the and dorsal striatum (Deng et al., 1999). Together, these observations suggest that c-fos induction after injections of toxic METH doses might occur to promote the induction of protective mechanisms such as the production of antioxidant enzymes or BDNF to attenuate METH neurotoxicity.
Further support for a role of IEGs in METH-induced cell death was provide by Jayanthi et al. (2005) who found that increased expression of expression of members of the Jun, Egr, and Nur77 subfamilies of transcription factors (TFs) occurred concurrently with increased markers of cell death in the rodent brain. They found, in addition, that these increases were accompanied increased expression of Fas ligand (FasL) mRNA which is known to be regulated by several IEG transcription factors. Moreover, METH neurotoxicity was accompanied by increased FasL protein expression in striatal GABAergic neurons that express enkephalin. There was also METH-induced cleavage of caspase-3 in FasL- and Fas-containing neurons. Importantly, pre-injections of the dopamine D1 receptor antagonist, SCH23390, that block the METH-induced IEG and FasL responses also attenuated METH-induced neuronal apoptosis.
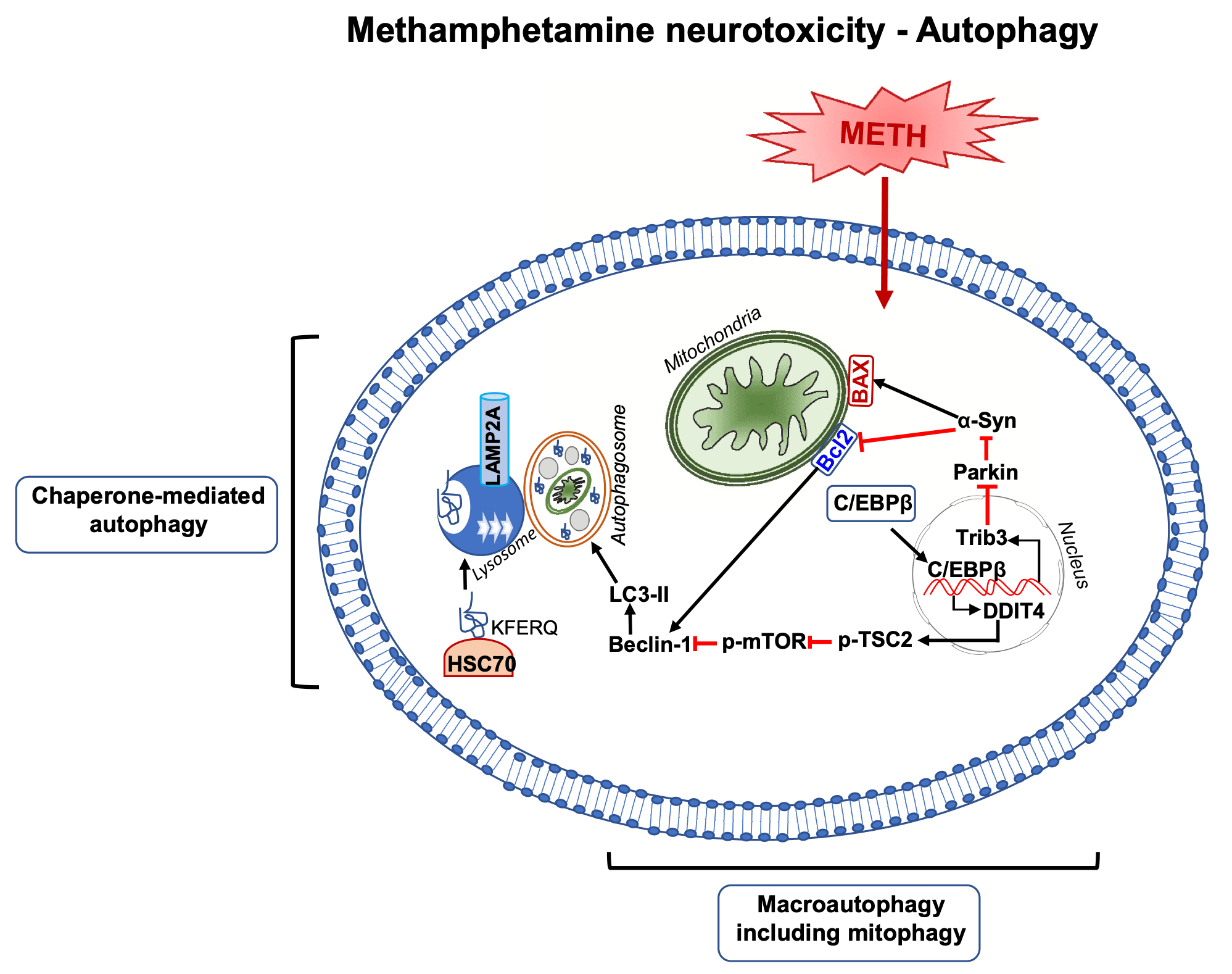
7. Meth neurotoxicity and autophagy
Autophagy plays an important role in the maintenance of neuronal function (Yamamoto and Yue, 2014; Ariosa and Klionsky, 2016). Autophagic processes are highly conserved with multiple ordered sequences of events that are tightly regulated by autophagy-related proteins (Klionsky, 2007; Mizushima, 2007; Meijer and Codogno, 2004). The sequences are initiated via the formation of a phagophore that can be triggered by a diversity of cellular stressors that include food or energy deprivation and hyperthermia (Klionsky and Emr, 2000; Cuervo et al., 2004; Chu, 2008). This initial step is regulated by the phosphoinositide 3-kinase (PI3K)-Beclin-1-Atg14-Vps15 complex (Klionsky and Emr, 2000; Klionsky, 2007). A subsequent step involves the formation of the autophagosome, a process by which the phagophore expands and engulfs the cytosolic component controlled by autophagy-associated genes (Atg) genes through Atg12-Atg5 and LC3 complexes (Sánchez-Martín and Komatsu, 2020). Ultimately, the fusion of autophagosomes and lysosomes leads to degradation of cytosolic contents (Gatica et al., 2018; Nakamura and Yoshimori, 2017). Autophagic mechanisms are thought to participate in molecular and biochemical pathways that modulate neurodegenerative processes that constitute the substrates of diseases such as Alzheimer’s and Parkinson’s disease (Giorgi et al., 2021; Lu et al., 2020). It was therefore of interest to investigate the effects of METH on autophagic mechanisms.
METH-induced autophagic changes were initially reported by Larsen et al. (2002) who document the formation of autophagic granules upon exposure to the drug. Castino et al. (2008) also observed that METH can also cause autophagosome formation in a cell culture system. Of note, genetic inhibition of autophagy in rat dopaminergic has been reported to exacerbate METH-induced apoptosis (Lin et al., 2012), thus implicating autophagic mechanisms as protective factors against METH neurotoxicity. Overexpression of LC3-II (microtubule-associated light chain 3), an autophagy regulatory protein, was also shown to protect against METH-induced cell death (Lin et al., 2012). Those results are not consistent with those of other investigators who have shown that inhibition of autophagy via mTOR(negative regulator of autophagy) attenuated METH-induced cell death (Kongsuphol et al., 2009; Li et al., 2012). Moreover, Xu et al. (2018) have intimated that autophagy may constitute an early response in a METH-induced cell death cascade. Xu et al. (2018) injected high doses of METH and reported increased protein expression of autophagy markers, Beclin-1 and LC3-II, in the rat striatum (Xu et al., 2018). A recent study by Subu et al. (2020)had also reported the observation that rats that self-administered large quantities of METH during a self-administration experiment exhibited significant alterations in markers of autophagy and neuronal apoptosis in their dorsal striatum.
Other investigators have also provided evidence for METH-induced autophagy. For example, Li et al. (2016) also reported that METH exposure increased the expression of DNA damage-inducible transcript 4 (DDIT4) and upregulation of Beclin-1 and LC-II. Yang et al. (2019)showed that large doses of METH can increase Beclin-1 expression in human neuroblastoma cells via the AKT- mTOR signaling pathway. Both AKT and DDIT4 are negative mTOR regulators that promote the formation of autophagosomes (Moore et al., 2016; Miao et al., 2020; Wang et al., 2012). A recent study by Huang et al. (2019) also documented METH-induced increased expression of tribbles homolog 3 (Trib3), an inducible ER stress protein (Ohoka et al., 2005), that participates in autophagic mechanisms (Ord and Ord, 2017). Trib3 was shown to decrease p-Akt/p-mTOR interaction that resulted in increased expression of Beclin-1 and LC3-II (Huang et al., 2019).
Chaperone-mediated autophagy (CMA) (Dice, 2007) has also been reported after toxic METH doses (Sun et al., 2019). CMA is different from micro- or macroautophagy in that degradation can occur with vesicle formation (Dice, 2007). CMA degrades thirty percent of cytosolic proteins during prolonged nutrient deprivation (Dice, 2007). Molecular chaperones in the cytoplasm and within lysosomes are responsible for this degradation pathway. One of the critical CMA components is the lysosome-associated membrane protein (LAMP) type 2A, a receptor located on the lysosomal membrane (Cuervo and Dice, 1996). LAMP-2A is the rate limiting step for CMA (Cuervo and Dice, 1996). Another protein of interest is the heat shock protein of 70kd (hsc70) which can form a complex with LAMP-2A (Dice, 2007). METH exposure was accompanied by time- and dose-dependent increases in LAMP-2A expression in human neuroblastoma cell lines, PC12 cells, and primary mice neurons (Sun et al., 2019). Interestingly, LAMP-2A silencing exacerbated METH-induced cell death (Sun et al., 2019), suggesting that CMA might work as a protective mechanism in those in vitro models. Much more remains to be done to understand the potential role of CMA in METH-induced neurotoxicity using in vivo models.
Interestingly, C/EBPβ, appears to be a critical effector of METH-induced autophagy via the activation of DDIT4/TSC2/mTOR signaling or Trib3/Parkin/alpha-synuclein mechanisms (Huang et al., 2019; Xu et al., 2018). A report by Li et al. (2017) has also suggested a role for glycogen synthase kinase3β (GSK3β) in METH-induced autophagy and neurodegeneration via the promotion of Tau and α-syn phosphorylation, α-syn accumulation, inhibition of lysosomal degradation, and consequent apoptotic cell death.
8. METH neurotoxicity and potential relevance to therapeutic approaches
METH users have been reported to suffer from cognitive impairments that can impact their activities of daily living and course of treatment when they seek treatment (Cadet and Bisagno, 2015). Importantly, there is a suggestion that recovery of cognitive functions can occur in conjunction with improvement in impulsivity and self-regulation after a psychological intervention with working memory training during in-patient treatment for methamphetamine use disorder (Brooks et al., 2016). These observations are supported by pre-clinical studies that have reported improvement in cognitive functions after administration of pharmacological compounds such as ZSET1446, a T-type calcium channel activator (Ito et al., 2007), silibinin, a natural polyphenolic flavonoid (Lu et al., 2010), 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl) benzamide (CDPPB) a mGluR5 allosteric modulator (Reichel et al., 2011) and modafinil, a dopamine uptake blocker (Kalechstein et al., 2010; González et al., 2014; Reichel et al., 2014). The ameliorating effect of ZSET1446 (azaindolizinone derivative, a T-type calcium channel activator) and modafinil (2-[(diphenylmethyl) sulfinyl] acetamide) on METH-induced impairment of recognition memory is mediated via activation of ERK cascade (Ito et al., 2007; González et al., 2014). In addition to cognitive improvement, modafinil can also provide protection against METH-induced cell death and neuroinflammation in the rodent models (Raineri et al., 2012), it is a drug that might have substantial anti-neurotoxic effects in humans who are treated with it. Silibinin mediates its ameliorating effects by reducing dopamine and serotonin levels in the prefrontal cortex and hippocampus, respectively (Lu et al., 2010). CDPPB, a positive allosteric modulator of mGluR5 receptors, improved METH-induced cognitive impairmentsvia interactions with these receptors (Reichel et al., 2011).
Pharmacological interventions that target ROS and RNS signaling cascades also hold great potential in alleviating METH-induced neurotoxicity. Specifically, selenium, a dietary antioxidant is known to reduce METH-induced ROS production (Kim et al., 1999; Imam et al., 1999). Another antioxidant, N-acetyl-L-cysteine, can also suppress METH-induced oxidative stress in both rodent (Zhang et al., 2012) and primate (Hashimoto et al., 2004) models. Moreover, inhibitors of nitric oxide synthase, (7-nitroindazole and AR-R17477AR), and a selective peroxynitrite scavenger, (5,10,15,20-tetrakis [2,4,6-trimethyl-3,5-sulfonatophenyl] porphyrinato iron III (FeTPPS)), have been shown to reduce METH-induced hyperthermia, peroxynitrite production, and METH-induced dopaminergic depletion (Imam et al., 2000; Sanchez et al., 2003). Although much remains to be done to test whether these agents could improve cognitive functions in METH-treated animals, when taken together, these results suggest that the addition of anti-oxidative compounds may improve cognitive by reducing the neurotoxic effects of METH.
9. Conclusions
This review has discussed the evidence that has documented the toxic effects of METH in the central nervous system. These include degeneration of monoaminergic terminals and neuronal apoptosis. There is now evidence that METH administration is also accompanied by autophagic changes in the brain. It remains to be clearly determined if these autophagic changes are precursors of neuronal apoptosis or serve as attempts to protect the neurotoxicity of the drug. We have provided a schema (Fig. 1) that illustrates the various biochemical cascades that have been shown to work as an ensemble to cause terminal damage and neuronal apoptosis in the mammalian brain. Although much more remains to be done to document if autophagy occurs in the brains of human METH users, a recent paper has reported that METH users exhibit increased Atg5 and LC3 in the pre-frontal cortical region (Khoshsirat et al., 2020). Nevertheless, because there is evidence that there might be pathobiological events in the brains of METH users who consume large quantities of the drug, it is essential that developers of pharmacological agents to treat METH use disorder take these toxic consequences into consideration. (See Fig. 2, Fig. 3, Fig. 4.) (See Table 1.)