Abstract
Alcohol addiction is a chronic relapsing brain disease characterized by an impaired ability to stop or control alcohol use despite adverse consequences. A main challenge of addiction treatment is to prevent relapse, which occurs in more than >50% of newly abstinent patients with alcohol disorder within 3 months. In people suffering from alcohol addiction, stressful events, drug-associated cues and contexts, or re-exposure to a small amount of alcohol trigger a chain of behaviors that frequently culminates in relapse. In this review, we first present the preclinical models that were developed for the study of alcohol seeking behavior, namely the reinstatement model of alcohol relapse and compulsive alcohol seeking under a chained schedule of reinforcement. We then provide an overview of the neurobiological findings obtained using these animal models, focusing on the role of opioids systems, corticotropin-release hormone and neurokinins, followed by dopaminergic, glutamatergic, and GABAergic neurotransmissions in alcohol seeking behavior.
Abbreviations
P rats: Indiana alcohol Preferring rats
CPP: conditioned place preference
MOP: Mu-opioid receptor
NAc: nucleus accumbens
OFC: orbitofrontal cortex
KOP: kappa-opioid receptor
DOP: delta-opioid receptor
NOP: nociceptin receptor
DYN: dynorphin
nor-BNI: nor-binaltorphimine
CRH: corticotropin-releasing hormone
BNST: bed nucleus of the stria terminalis
JNK: c-Jun N-terminal kinase
N/OFQ: nociceptin/orphanin FQ
CeA: central nucleus of the amygdala
GPCRs: G protein-coupled receptors
HPA: hypothalamic-pituitary-adrenal
SP: substance P
NK: neurokinin
VTA: ventral tegmental area
(m)PFC: (medial) prefrontal cortex
MSNs: medium spiny projection neurons
ADE: alcohol deprivation effect
COMT: Cathechol-O-Methyltransferase
DLS: dorsolateral striatum
iGluR: ionotropic glutamate receptors
mGluR: metabotropic glutamate receptors
BLA: basolateral amygdala
DMS: dorsomedial striatum
NMDAR: N-methyl-d-aspartate receptors
AMPAR: α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors
OFC: orbitofrontal cortex
PAM: positive allosteric modulator
CaMKII: Ca2+/calmodulin-dependent protein kinase II
NAM: negative allosteric modulator
MPEP: 2-Methyl-6-(phenylethynyl)pyridine
ERK1/2: extracellular signal-regulated kinases ½
CDPPB: 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide)
LH: lateral hypothalamus
mIPSC: miniature inhibitory postsynaptic current
sP: sardinian alcohol preferring
1 INTRODUCTION
A main challenge of addiction treatment is to prevent relapse after patients achieve abstinence. Half a century ago, it was reported that more than 50% of newly abstinent patients with alcohol addiction (hereafter equated with alcoholism, alcohol dependence, or moderate – severe alcohol use disorder) relapse within three months (Hunt et al., 1971). Disappointingly, these numbers have remained largely unchanged over time (Sinha, 2011). In people suffering from alcohol addiction, stressful events, drug-associated cues and contexts, or re-exposure to a small amount of alcohol (“priming”, or “the first drink”) trigger a chain of behaviors that frequently culminates in relapse (Brownell et al., 1986; Hendershot et al., 2011). An urge to drink, or “craving”, is often (but not always) an antecedent of relapse (Wray et al., 2014). Its causal role for initiating substance use has long been debated (Tiffany, 1990), but research has shown that the magnitude of craving in response to triggers, assessed under controlled laboratory conditions, reliably predicts the risk of relapse in the subsequent months (Sinha et al., 2011).
1.1 Animal models for the study of alcohol seeking behavior
1.1.1 Alcohol seeking in reinstatement models to study craving and relapse
Since its introduction in a seminal study (de Wit & Stewart, 1981), reinstatement of drug seeking following extinction has become the most common model to study relapse in animals, and to investigate the underlying neural mechanisms (Epstein et al., 2006). To reinstate alcohol-seeking, it is first necessary to initiate robust and stable levels of alcohol self-administration. Operant self-administration, a workhorse of addiction research, poses some unique challenges when applied to alcohol. One of these is the aversive taste of high alcohol concentrations for most rodents, a phenomenon shared by humans with little or no experience of alcohol (Koob et al., 2003). Widely used protocols to overcome this barrier have required water deprivation, saccharin/sucrose fading (Samson, 1986; Samson et al., 1988), pre-exposure to alcohol in the homecage, or extended training to initiate the acquisition and maintenance of self-administration (Simms et al., 2010). Although effective, these procedures introduce a potential for confounds in alcohol self-administration studies, and even more so when examining reinstatement of responding. More recently, work from our lab and others has shown that robust and stable levels of alcohol self-administration can be achieved without resorting to these approaches (Augier et al., 2014, 2017; Puaud et al., 2018).
BOX . Mini-Dictionary of Terms
Operant self-administration
A procedure in which an animal is trained to perform an operant response (most of the time, pressing a lever or nose-poking) to obtain a reward, usually food pellets, or a drug solution that can be delivered orally in a drinking spout or intravenously depending of the drug studied and the method of administration chosen by the experimenter. In the vast majority of conditioning experiments, two levers (or two holes) are presented. Pressing on the "active" lever allows the animal to obtain the reward, whereas responding on the "inactive" lever has no behavioral consequences. This learning procedure is based on operant or Skinnerian conditioning. If the animal is able to learn the response/reward association and repeat it, its behavior is considered as reinforced and the drug is a reinforcer.
Reinstatement
The gold standard of animal models to study drug relapse. Following drug self-administration acquisition, maintenance and subsequent extinction of the drug-associated responding, animals are tested for reinstatement of their drug seeking behavior induced by different kind of stressors (pharmacological, physical, and psychological), drug-priming, discrete cues or contextual cues.
Stress-induced reinstatement
In this variant of the reinstatement paradigm, laboratory animals are initially trained to self-administer a drug, for which delivery is paired with discrete cues (tone, cue light, noise of the injection pumps, smell of the alcohol solution). Operant responding (lever presses or nose pokes) is then extinguished in the presence of the drug-associated discrete cues.
Cue-induced reinstatement
Similarly to stress-induced reinstatement, animals are first trained to self-administer a drug in the presence of concomitant discrete cues. Their responses are consecutively extinguished in the absence of the cues previously associated with the drug. During reinstatement testing, reintroduction of these discrete cues precipitate relapse-like behavior as shown by increased responses on the lever associated with the drug.
Drug-induced reinstatement
Animals are similarly trained to self-administer a drug and drug delivery is paired with a discrete cue. Operant responding is then extinguished in the presence of the discrete cues. Once stable and low rate of responses is achieved, responding for the drug is reinstated by a unit dose of the drug previously self-administer (drug priming).
Once alcohol self-administration is acquired, the reinstatement procedures start with an extinction phase, in which the operant response that previously led to an alcohol delivery no longer has a programmed consequence. Following extinction training, responses on the alcohol-associated lever decrease to low levels or stop. Reinstatement of responding for alcohol under extinction conditions (i.e. in the absence of the reinforcer) can then be induced by triggers, with discrete cues and stress being most robust for alcohol (Figure 1). The rate of operant responding (i.e reinstatement) on the lever previously associated with alcohol delivery is taken as a measure of the animal's urge to obtain alcohol, a model of craving in patients.
FIGURE 1. Animal models for the study of alcohol seeking behavior.
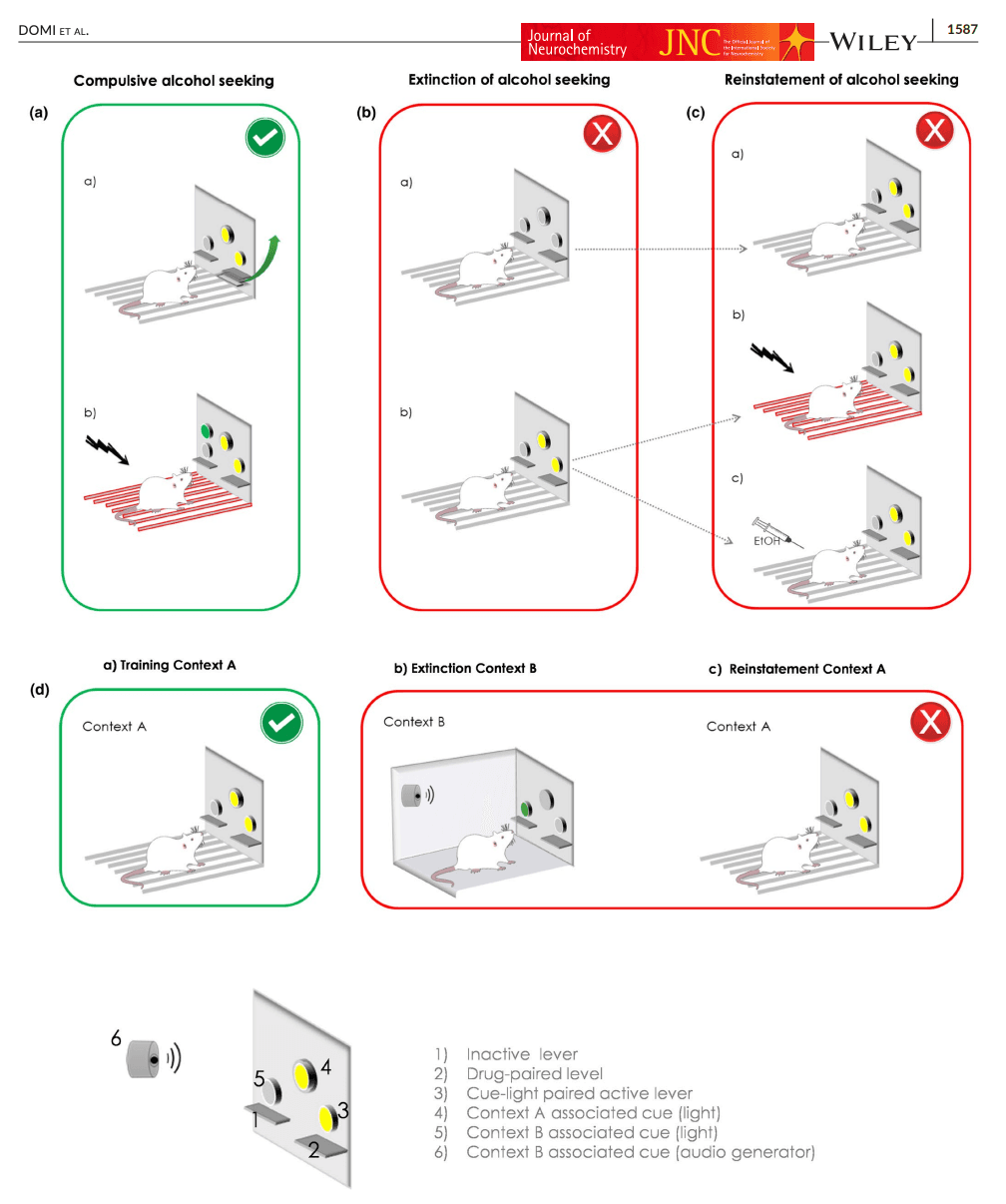
Priming injections of alcohol can successfully reinstate responding in rats (Le et al., 1998), but are less commonly used. With alcohol, reinstatement is more robustly produced by discrete alcohol-associated cues (Sinclair et al., 2012) or contexts (Chaudhri et al., 2009; Hamlin et al., 2009) but the efficacy of these stimuli to trigger reinstatement may rely on the specific type of cue presented. For instance, exposure to olfactory but not auditory cues can trigger relapse in rodents (Katner et al., 1999). In addition, reinstatement of alcohol seeking was potentiated when the olfactory cue was combined to a discriminative visual cue, enduring resistance to extinction, specifically in genetically selected alcohol preferring P rats (Ciccocioppo et al., 2001). A robust alcohol reinstatement is also produced by physical stressors such as intermittent footshock (Le et al., ,1998, 2002; Le & Shaham, 2002) or pharmacological stressors such as the anxiogenic drug yohimbine (Cippitelli et al., 2010; Le et al., 2005). For example, Le and co-workers found in their seminal study that exposure during 5 and 15 min to intermittent footshock (0.5 s shock, intensity of 0.8 mA) potently reinstated responding for alcohol, but not sucrose. By contrast, a priming injection of a dose of 0.48 g/kg of alcohol only marginally reinstated responding (Le et al., 1998).
However, because stressors that precipitate relapse in patients with alcohol addiction are typically psychosocial, they differ from the type of triggers used in preclinical reinstatement studies. This may be a limitation (Bjorkqvist, 2001; Epstein et al., 2006; Katz & Higgins, 2003), and it cannot be excluded that molecular mechanisms identified in reinstatement studies may differ from those that promote relapse in humans for this reason. In an attempt to address this issue, a recent study used the resident-intruder paradigm to study the effect of social defeat stress on cocaine seeking (Manvich et al., 2016). When rats were re-exposed to cues predictive of psychosocial stress (olfactory cues that signaled sessions of defeat stress), they potently reinstated lever pressing for cocaine. Whether this observation would generalize to reinstatement of alcohol-seeking, and whether the neural mechanisms mediating reinstatement behavior differ between these types of stressors is an important question for future studies.
Although used less commonly, reinstatement can also be assessed using conditioned place preference (CPP) procedures (Mueller & Stewart, 2000), or the operant runway model of drug-self administration (Ettenberg et al., 1996; Geist & Ettenberg, 1990). These procedures have to our knowledge not been applied to alcohol seeking.
1.1.2 Compulsive alcohol seeking
Alcohol seeking and taking that becomes “compulsive”, i.e. continues despite negative consequences, is a hallmark of alcohol addiction (Corbit et al., 2012; Everitt & Robbins, 2016; Koob & Volkow, 2010; Wagner & Anthony, 2002). Understanding the transition from controlled to compulsive alcohol use is a critical challenge for addiction research. Most preclinical alcohol studies have focused on compulsive alcohol taking, as assessed by the persistence of animals to drink alcohol despite adulteration with the bitter tastant, quinine (Wolffgramm, 1991; Wolffgramm & Heyne, 1995), or more recently, their persistence to self-administer alcohol in operant procedures despite adverse consequences such as an electric footshock delivered contingently with the alcohol (Augier et al., 2018; Seif et al., 2013). Compulsive alcohol seeking in the absence of alcohol, preceding actual intake (Everitt & Robbins, 2005) has only recently begun to be studied.
In a recent paper, the authors adapted procedures previously developed to study cocaine-seeking in rats (Pelloux et al., 2007; Vanderschuren & Everitt, 2004), and used these to disentangle alcohol seeking from alcohol taking using Indiana alcohol-preferring P rats (Giuliano et al., 2018, 2019).
The protocol used to study compulsive seeking in these experiments can be divided into four main phases (Figure 1a). First, rats undergo Pavlovian conditioning (1), in which they are trained to associate a 20s cue-light, which serves a conditioned stimulus, with the availability of a 15% alcohol solution. Next, during the taking phase (2), one of the two levers is randomly assigned as a “taking lever”, and operant responses on this lever are reinforced with the delivery of alcohol, together with presentation of the cue light. During the seeking-taking phase (3), the other lever serves as the “seeking lever”, and operant responses on this lever under a random interval schedule (with interval length progressively increased from 5 to 60 s) lead to the presentation of the taking lever, while the seeking lever is retracted. Similar to phase ii, pressing on the taking lever is now reinforced with a delivery of alcohol, together with activation of the cue light. After this, both levers retract, and rats need to re-initiate this chained schedule of reinforcement in order to drink more alcohol. Finally, during the last phase (4), the seeking-taking chain schedule becomes punished. Rats now receive mild footshocks (0.25 increased to 0.45 mA over daily sessions), randomly delivered on 30% of the trials. Using this protocol, a cluster analysis identified three subgroups of rats. Following the introduction of unpredictable punishment associated with the seeking lever, 34% of the population showed punishment-resistant alcohol seeking, whereas 30% of the rats markedly reduced their responses on the seeking lever. The rest of the animals (36%) were classified as intermediate, and partially suppressed alcohol seeking behavior (Giuliano et al., 2018).
Finally, an alternative approach to study both unpunished and punished alcohol seeking in preclinical models has been provided by multicriteria paradigm. Based on the seminal work of Deroche-Gamonet and co-workers (Deroche-Gamonet et al., 2004), alcohol seeking has been recently studied in a multisymptomatic addiction model that characterizes addiction-prone phenotype in rats, derived from the DSM-IV/5 diagnostic criteria of addiction (Domi et al., 2019; Jadhav et al., 2017). Alcohol-seeking was measured during “no-drug” periods as a progressive daily increase in seeking when responding for alcohol was neither reinforced by conditioned stimuli nor alcohol delivery. Over time, only one-third of rats developed persistence in alcohol seeking. In one of these papers (Domi et al., 2019), alcohol seeking despite a punishment was also assessed. Rats were punished with a 0.3 mA footshock that anticipated alcohol taking response. Punishment-resistant alcohol seeking was observed only in a subset of individuals, confirming the inter-individual vulnerability to develop alcohol addictive behaviors.
2 NEUROBIOLOGICAL MECHANISMS MEDIATING ALCOHOL SEEKING
2.1 Section I: opioid systems and alcohol seeking
In reviewing the neurobiology of alcohol seeking, opioid systems offer a useful starting point, since the opioid antagonist naltrexone and its structural analog nalmefene are clinically approved treatments for alcohol addiction. Meta-analysis of randomized controlled trials robustly show that naltrexone reduces relapse to heavy drinking (Jonas et al., 2014; Mann et al., 2013). In contrast with acamprosate, naltrexone is not effective for maintaining abstinence. Its clinical profile thus indicates an ability to block the progression from a slip, in which alcohol is sampled, to relapse and heavy drinking. This closely parallels blockade of priming-induced reinstatement in rats, which has also been reported with naltrexone (Le et al., 1999). Furthermore, meta-analysis of human laboratory studies supports an ability of naltrexone to suppress cue-induced craving (Hendershot et al., 2017). This is presumably related to the observation that, in patients with alcohol addiction, elevations of striatal mu-opioid receptors (MOP) correlate with subjective cravings 1 to 3 weeks into abstinence (Heinz et al., 2005), while, in social drinkers, alcohol administration results in release of endogenous opioids in the nucleus accumbens (NAc) and orbitofrontal cortex (OFC) (Mitchell et al., 2012). MOP activation is likely to promote and modulate alcohol-induced dopamine release in humans (Ramchandani et al., 2011). These and other data provide some degree of support for a predictive validity of preclinical reinstatement models, and may offer opportunities for reverse-translational research strategies (Venniro et al., 2020). It is, however, clear that the degree of predictive validity of these models may vary on a system-by-system basis (see below).
Opioid systems comprise a vast and complex landscape of neuropeptide ligand families (endorphins, dynorphins, enkephalins, and the related non-opioid peptide nociceptin), as well as their receptor families [MOP, kappa-opioid (KOP), delta-opioid (DOP) receptors; the nociceptin receptor (NOP), while related, does not bind to opioid ligands]. Opioid systems, and their multitude of roles in addictive disorders have been the subject of multiple excellent reviews [e.g. (Lutz & Kieffer, 2013)], including reviews that have specifically summarized the role of opioid systems in alcohol addiction (Nutt, 2014). Opioid receptors are Gi-coupled, highly expressed in brain areas of importance for reinforcing properties of drugs, and are involved in the regulation of both unconditioned and conditioned behavioral effects of alcohol. For instance, stimulation of MOP regulates the positive reinforcing effects of alcohol, whereas activation of KOP mediates aversive and dysphoric aspects of alcohol effects. In the following, we describe the involvement of the opioid receptor subtypes with a focus on alcohol seeking in laboratory animals (see Table 1 for a summary).
TABLE 1. Compounds targeting the opioid system in alcohol seeking behavior
2.1.1 MOP receptors and alcohol seeking
In a parallel to human findings, the non-selective opioid antagonist, naltrexone was first shown to suppress reinstatement of alcohol seeking triggered by a priming dose of alcohol (Le et al., 1999). In a further parallel to human observations, MOP blockade results in reduced incentive motivational (anticipatory) responses for alcohol and reinstatement of alcohol seeking by alcohol-associated stimuli. Naltrexone, at doses that are likely to predominantly act through MOP blockade (0.25, 1.0 mg/kg), selectively inhibits cue- but not stress-induced reinstatement in Wistar rats (Liu & Weiss, 2002). It has also been reported that both naltrexone and the selective of MOP antagonist, naloxonazine, (1–15 mg/kg) inhibits cue-induced alcohol-seeking (Ciccocioppo et al., 2002). In agreement with findings in rats, naltrexone (0.32–3.2 mg/kg) has also been shown to reduce motivation to drink in the presence of alcohol-related cues in baboons (Kaminski et al., 2012). On the other hand, naltrexone, even when given at higher doses (3 and 10 mg/kg) during extinction had minimal effects on subsequent sensitivity to alcohol cues and alcohol consumption (Ciccocioppo et al., 2002). Furthermore, naltrexone given during repeated alcohol cue exposure does not alter the subsequent incentive value of alcohol cues presented in its absence, or enhance exposure-induced extinction, a procedure that parallels clinical cue-exposure therapy (Williams & Schimmel, 2008).
The selective and potent MOP antagonist GSK1521498 has been shown to reduce both alcohol drinking and cue-induced alcohol seeking in alcohol-preferring P rats (Giuliano et al., 2015). GSK1521498 has also been evaluated in a model of compulsive alcohol seeking that relies on a chained seeking-taking schedule (see Section 1.1.2). GSK1521498 reduced alcohol seeking under non-punished conditions both in rats previously identified as compulsive, and those that were not. However, the degree with which seeking behavior was suppressed was greater in the compulsive rats, potentially suggesting that the therapeutic value of GSK1521498 may be particularly pronounced in individuals with a higher degree of alcohol addiction severity (Giuliano et al., 2018). In contrast with naltrexone, GSK1521498 is selective for MOP, and lacks partial agonist activity (Nathan et al., 2012).
Collectively, preclinical data strongly support MOP-blockade as a treatment to prevent alcohol craving and relapse, in agreement with clinical findings. Clinical effect sizes achieved through this mechanism are, however, modest (Del Re et al., 2013). It is unclear whether more selective MOP antagonists have a potential to improve outcomes beyond what is achieved with currently approved medications, since near complete MOP blockade can be achieved using naltrexone (Lee et al., 1988), and the duration of occupancy can be further improved using nalmefene (Ingman et al., 2005).
2.1.2 KOP receptors and alcohol seeking
KOPs and their endogenous ligand dynorphin (DYN) play a critical role in stress-reactivity and negative emotionality in addictive disorders, including alcohol addiction (Bruchas et al., 2010). Prolonged alcohol exposure in rats induces long-term neuroadaptation in the KOP/DYN system, resulting in negative affective-like states that promote excessive drinking presumably through negative reinforcement (Walker & Koob, 2008). Increased KOP sensitivity, together with a hypodopaminergic state in the NAc, is a key mechanism in mediating the aversive properties of alcohol withdrawal (Rose et al. 2016), because an increased activity of DYN/KOP during protracted abstinence may contribute to a negative emotional state that facilitates alcohol seeking, KOP antagonists may have a potential to become useful therapeutics in alcohol addiction (Drews & Zimmer, 2010).
In rats, the prototypical KOP antagonist nor-binaltorphimine (nor-BNI) has been shown to suppress stress-induced alcohol seeking by both a physical stressor (footshock) and the pharmacological stressor yohimbine (Funk et al., 2014; Harshberger et al., 2016). Conversely, activation of KOP receptors using systemic administration of the prototypical KOP agonist U50,488 reinstates alcohol seeking, and this is blocked by nor-BNI. Reinstatement triggered by KOP activation in this study was also blocked by pretreatment with the corticotropin-releasing hormone type-1 (CRH1; see Section 2.2.1) receptor antagonist antalarmin, indicating that DYN acts upstream of CRH to produce stress-induced reinstatement (Funk et al., 2014). nor-BNI has also been shown to reduce U50,488-induced reinstatement of alcohol seeking when injected in the bed nucleus of the stria terminalis (BNST; 4 μg/side) in Long Evans rats, suggesting BNST as a key player in DYN/KOP mechanisms of stress-induced alcohol seeking (Crowley et al., 2016; Erikson et al., 2018; Le et al., 2018).
The KOP antagonist nor-BNI has also been shown to block cue-induced reinstatement of alcohol seeking (Funk et al., 2014). We have also previously shown that JDTic (Carroll et al., 2004), a first-generation non-peptide selective KOP antagonist, blocked withdrawal-induced anxiety-like behavior and reduced cue-induced reinstatement in Wistar rats (Schank et al., 2012). In another study, conducted in female alcohol preferring P rats, JDTic dose dependently (1, 3, or 10 mg/kg) reduced relapse-like responding tested in the Pavlovian Spontaneous Recovery test (Deehan et al., 2012). Because of the complex actions of nor-BNI and JDTic discussed below, it is unclear how these effects are related to their acute KOP blockade.
Based on these and other findings, KOP blockade has been considered as a mechanism with therapeutic potential in alcohol addiction, but KOP antagonists with properties making them suitable candidates for clinical development have been lacking. Presumably because of phosphorylation of c-Jun N-terminal kinase (JNK), nor-BNI has effects that last long after it has dissociated from the receptor, resulting in complex pharmacokinetic and pharmacodynamic properties. JDTic has a similarly complex pharmacology, related to non-competitive effects likely to be mediated by modulation of JNK signaling (Bruchas et al., 2007), and was terminated from clinical development because of cardiac toxicity (Buda et al., 2015). A new generation of KOP antagonists may finally allow their evaluation for clinical efficacy. A representative of this generation is CERC-501 (Rorick-Kehn et al., 2014), a novel and a selective KOP antagonist that has been found safe in Phase 1 in both healthy and cocaine dependent subjects (Lowe et al., 2014; Naganawa et al., 2016; Reed et al., 2017).
We have evaluated CERC-501 in a battery of preclinical tests to assess its potential as a clinical candidate in alcohol addiction (Domi et al., 2018). At an oral dose of 10 mg/kg, CERC-501 fully reversed the anxiogenic effects of alcohol withdrawal, and blocked stress-induced reinstatement of alcohol seeking. These effects were highly specific behaviorally, since the same dose did not affect cue-induced reinstatement or nicotine induced escalated-drinking (Domi et al., ,2020, 2021). These findings are in agreement with the hypothesis that KOP activation is primarily associated with negative emotional states, and their ability to promote alcohol seeking and taking. The profile of CERC-501 complements that of naltrexone, which selectively inhibits cue-induced but not stress-induced reinstatement (Liu & Weiss, 2002). Combining KOP and MOP antagonism in clinical treatment therefore appears to be an attractive strategy. The preclinical safety profile of CERC-501 is promising for clinical development, since it did not affect the sedative properties of alcohol, its metabolism or general locomotor activity.
2.1.3 DOP receptors and alcohol seeking
In contrast with the rich literature on MOP and KOP, few studies have characterized the role of DOP on alcohol seeking behavior. Some data suggest that DOP may be involved in cue-induced reinstatement of alcohol-seeking. For example, the δ selective antagonist naltrindole, at a dose of 5 mg/kg (i.p.) selectively inhibited alcohol-seeking induced by alcohol-related environmental stimuli (Ciccocioppo et al., 2002). In agreement with this result, naltrindole, but not the MOP antagonist CTOP potently suppressed both cue-induced and context-induced reinstatement of alcohol seeking (Marinelli et al., 2009). Finally, the DOP antagonist SoRI-9409 effectively and dose-dependently reduces yohimbine stress-induced reinstatement of alcohol-seeking in rats (Nielsen et al., 2012).
2.1.4 NOP receptors in alcohol seeking
Nociceptin/orphanin FQ (N/OFQ), a 17 amino acid peptide, is the endogenous ligand for the NOP. The N/OFQ-NOP system is involved in modulation of pain processing, affective states, and other physiological functions such as neuroendocrine and immune response (Bodnar, 2013; Valentino & Volkow, 2018). It has also been the subject of extensive investigation in models of addictive disorders, including alcohol. Originally, the overarching hypothesis was that NOP activation attenuates multiple measures of motivation for addictive drugs. This hypothesis has subsequently become complicated by observations that similar effects are also produced by NOP antagonists, making it unclear whether agonists or antagonists are most likely to offer opportunities to develop medications for addictive disorders (Ciccocioppo et al., 2019).
NOP activation, whether by nociceptin itself, peptide analogues, or small-molecule non-peptide agonists, has been shown to reduce expression of alcohol withdrawal signs, relapse after alcohol deprivation, and stress-induced reinstatement of alcohol seeking. This has been observed both in non-dependent Wistar rats, and, to an even higher extent, following a history of dependence (Economidou et al., 2011; de Guglielmo et al., 2015; Kuzmin et al., 2007; Martin-Fardon et al., 2000). Central administration of nociceptin also suppressed cue-induced alcohol seeking in alcohol preferring msP rats (Ciccocioppo et al., 2004). Electrophysiological studies have shown that, in the central nucleus of the amygdala (CeA), alcohol induces more pronounced changes of the N/OFQ-NOP system in alcohol dependent and msP rats compared to non-selected, naïve rats (Herman et al., 2013). In addition, msP rats show an innate over-expression of CRH1 receptors, driven mainly by two single nucleotide polymorphisms at CRHR1 gene locus (Hansson et al., 2006). These findings provide an important link between the innate dysregulation of CRH with the N/OFQ-NOP system and excessive drinking (Martin-Fardon et al., 2010).
We have also shown that the potent, brain-penetrant small-molecule NOP agonist, SR-8993 (1, 3 mg/kg), is able to reverse acute alcohol withdrawal-induced anxiety, and attenuate both stress- and cue-induced relapse to alcohol seeking in Wistar rats (Aziz et al., 2016). Paradoxically, similar findings have been obtained with an orally available small-molecule NOP antagonist, LY2940094 (3, 10 mg/kg). Using this antagonist and alcohol preferring msP rats, it was shown that blockade of NOP can also prevent alcohol taking (Borruto et al., 2020) and stress-induced reinstatement to alcohol seeking. LY2940094 also blocked alcohol-induced dopamine release in the NAc (Rorick-Kehn et al., 2016).
In an attempt to reconcile these paradoxical findings, it has been hypothesized that NOP receptors undergo rapid desensitization in response to activation by agonists (Toll et al., 2016). This is potentially consistent with observations that systemic treatment with the NOP agonist, MT-7716, which suppressed both cue- and stress-induced reinstatement of alcohol, gradually reduced alcohol drinking with an effect persisting also after discontinuation of the drug (Ciccocioppo et al., 2014). Exogenous administration of NOP agonists may thus down-regulate NOP transmission through receptor desensitization, and result in an antagonist-like effect.
It is thus presently unclear whether targeting the NOP system is a fruitful avenue for developing alcohol addiction medications, and if so, whether agonists or antagonists would be the preferred strategy. Ultimately, human data are needed to provide answers to these questions. To date, the only human data available come from a small, 8-week proof-of-concept study with LY2940094. These are inconclusive, as the study was negative for its primary endpoint of number of drinks per day, but did show significant effects in several secondary analyses, including the objective biomarker of alcohol consumption, gamma-glutamyl transferase (Post et al., 2016).
2.2 Section II: other peptides involved in alcohol seeking
2.2.1 Corticotropin-releasing Hormone (CRH)
CRH, a 41 amino acid peptide best known for its role as the hypothalamic releasing factor for the adrenocorticotropic hormone, is also widely distributed outside the hypothalamus. Its biology and role in alcohol-related behaviors have been the subject of multiple reviews [e.g. (Heilig & Koob, 2007; Heinrichs & Koob, 2004; Zorrilla et al., 2013)]. In addition to high densities of CRH neurons within the paraventricular nucleus of the hypothalamus, CRH-positive cells are also present in structures involved in alcohol seeking, including CeA and BNST. Actions of CRH are mediated through two subtypes of Gs-coupled G protein-coupled receptors (GPCRs). Behavioral stress responses, including stress-induced alcohol seeking, are predominantly mediated by CRH1 receptors in CeA and BNST. Effects of CRH2 activation are less clear, but are commonly opposite to those of CRH1. Similar to many neuropeptide systems, CRH1 signaling that mediates behavioral stress responses is an “alarm system” that is quiescent under a wide range of conditions, but becomes activated in the presence of uncontrollable stress.
Blockade of CRH signaling robustly blocks stress-induced alcohol seeking, while leaving cue-induced relapse-like behavior unaffected. This was first demonstrated with intracerebral administration of the non-selective peptide antagonist D-Phe CRF12–41, as well as systemic administration of the selective small molecule CRH1 antagonist CP-154 526; both these approaches blocked stress-induced reinstatement of alcohol seeking. This study also demonstrated a central, hypothalamic-pituitary-adrenal (HPA) axis independent mediation CRH1 antagonism on stress-induced reinstatement, since adrenalectomy did not influence its ability to block reinstatement (Le et al., 2000). A subsequent study in rats with a history of alcohol dependence showed a dissociation between effects on stress- and cue-induced reinstatement, in which CRH antagonism was selective for reinstatement induced by stress (Liu & Weiss, 2002). Following a history of alcohol dependence, expression of CRH and its CRH1 receptor is up-regulated within the CeA (Sommer et al., 2008), and this is accompanied by a markedly increased sensitivity to blockade of stress-induced reinstatement by CRH antagonism (Gehlert et al., 2007).
Collectively, these and other data consistently show that in rodents, CRH1 receptors selectively mediate stress- but not cue-induced reinstatement, and that a recruitment of the CRH system following a prolonged history of alcohol dependence renders animals particularly sensitive to blockade of relapse-like behavior by CRH1 antagonism. This research predicted that preclinical findings with CRH1 antagonists would translate into suppression of stress-induced craving in people with alcohol addiction, an established biomarker that predicts clinical relapse (Sinha et al., 2011). The arrival of small-molecule CRH1 antagonists that were safe and well-tolerated in humans subsequently allowed an evaluation of this hypothesis.
Unfortunately, available studies do not find support for human translation of the preclinical findings (Kwako et al., 2015; Schwandt et al., 2016). These results may not be conclusive (Pomrenze et al., 2017; Shaham & de Wit, 2016), but it is noteworthy that both studies went to great length to ensure target engagement through the use of biomarkers, and that one of them used verucerfont, a “fast-on, slow-off” CRH1 type of CRH1 antagonist thought to be particularly effective to achieve a functional blockade of CRH1 receptors (Zorrilla et al., 2013). Combined with the failures of CRH1 antagonists on multiple other stress-related psychiatric indications (Binneman et al., 2008; Coric et al., 2010; Dunlop et al., 2017), and their termination in clinical development, it is in our view unlikely that this mechanism can be resurrected for treatment of alcohol addiction.
2.2.2 Substance P (SP) and its neurokinin 1 (NK1) receptor
SP is an 11 amino acid peptide that belongs to the tachykinin family, which also includes neurokinin A (NKA) and neurokinin B (NKB) (Pennefather et al., 2004). Tachykinins exert their effects through three receptor subtypes, NK1-3, among which SP preferentially binds to the NK1 receptor, while the NK2 receptors is preferentially activated by NKA, and the NK3 receptor by NKB. NK1 receptors are Gs/q-coupled GPCRs, are located in a range of brain regions involved in both appetitive and aversive behaviors, modulate behavioral responses to stress, and regulate several alcohol-related behaviors (Schank & Heilig, 2017).
A challenge for preclinical studies on the role of NK1 receptors is a limited sequence homology and ligand affinity profile between human and rodent NK1 receptors, which limits the utility of NK1 antagonists developed for human use for studies in rodents (Schank & Heilig, 2017). This was overcome through the synthesis of L822429, an NK1 antagonist specifically developed to possess high affinity at rat NK1 receptors (Ebner et al., 2004). Using this molecule as a tool, we found that systemic blockade of NK1 receptors blocks stress-induced reinstatement, an effect with high behavioral specificity, as the same dose of the antagonist left cue-induced reinstatement unaffected (Schank et al., 2011). In alcohol preferring P rats, NK1 expression in CeA is elevated because of a gene sequence variant enriched in this line (Schank et al., 2013). P rats show an increased sensitivity to reinstatement of alcohol seeking by the pharmacological stressor yohimbine, which is suppressed by intra-CeA infusion of L822429. Conversely, viral over-expression of NK1 receptors in the CeA of Wistar rats increases their sensitivity to yohimbine-induced reinstatement (Nelson et al., 2019). In addition, recent findings indicate that the role of NK1 receptors in promoting stress-induced alcohol seeking in the CeA may be related to the fact that activation of these receptors by SP increases GABA-release in the CeA, and that this effect is up-regulated following a history of dependence [(Khom et al., 2020); see Section 2.5]. Collectively, these findings show that NK1 receptors in the CeA promote sensitivity to stress-induced relapse, as well as other alcohol-related behaviors.
Based on preclinical findings, we evaluated the NK1 antagonist LY686017 in an academic experimental medicine study, carried out in recently detoxified patients with alcohol addiction. This study used stress-induced craving and brain responses to negative emotional stimuli as biomarkers, and found that LY686017 suppressed both (George et al., 2008). A subsequent Phase 2 study was carried out by Eli Lilly, and has not been published (NCT00805441). In contrast with the laboratory study, this study was carried out in unselected patients, who overall had a low level of anxiety, and was negative on the primary outcome. However, several secondary analyses suggested a signal for efficacy.
Development of NK1 antagonists was in part driven by the discovery of their potential as antidepressant medications (Kramer et al., 1998). Following inconsistent results in subsequent depression trials, development of NK1 antagonists for stress-related psychiatric disorders was discontinued throughout the pharmaceutical industry. It was only later that a key factor behind the inconsistent results was identified. In contrast with most GPCR antagonists, for which central receptor occupancy >90% is typically sufficient for therapeutic efficacy, robust effects of NK1 antagonists require a near complete blockade (Ratti et al., 2013; Rupniak & Kramer, 2017). In our view, it is therefore a possibility that NK1 antagonism remain a viable therapeutic mechanism in alcohol addiction, if delivered using a highly brain penetrant medication, administered at adequate doses, to anxious alcohol addicted patients. Unfortunately, this proposition may never be evaluated.
2.3 Section III: the role of dopaminergic neurotransmission in alcohol seeking
Alcohol activates dopaminergic neurons in the ventral tegmental area (VTA) resulting in increased dopamine release in forebrain cortico-limbic regions, including the NAc and medial prefrontal cortex (mPFC) (Di Chiara & Imperato, 1985; Ding et al., 2011; Gessa et al., 1985). The reinforcing effects of alcohol are in part dependent on this dopamine release, and inhibition of dopamine receptors reduces both alcohol self-administration (Ding et al., 2015; Engleman et al., 2020) and reinstatement of drug-seeking behaviors (Marinelli et al., 2003; McFarland et al., 2004). Alcohol-induced dopamine release also induces neuroplasticity which may further promote the development of addiction (Ma et al., 2018). Neuroplastic changes in reward- and memory-related circuits mediated by dopamine may further produce a hypofunctioning mPFC, resulting in diminished impulse control and increased vulnerability to drug relapse (Koob & Volkow, 2016; Langleben et al., 2008; Trantham-Davidson et al., 2014). Overall, the role of dopaminergic neurotransmission is dependent on the brain region studied, how long alcohol has been consumed, and the kind of alcohol seeking behavior that is monitored (see Table 2 for a summary).
TABLE 2. Compounds targeting the dopaminergic neurotransmission in alcohol seeking behavior
Dopamine elicited responses are mediated through five GPCRs. Based on sequence homology, and biological responses, these are divided into a D1-like and the D2-like family. The D1-like receptor family consists of dopamine D1 and dopamine D5 receptors, which share over 80% sequence homology within the transmembrane domains, but only 50% overall homology at the amino acid level (Sidhu, 1998). The D2-like family consists of dopamine D2, D3 and D4 receptors; the transmembrane regions of D3 and D4 receptors share 75% and 53% sequence homology with the D2 receptor, respectively (Sokoloff et al., 1992). The dopamine D1 and D2 receptors are the most abundant subtypes, and are highly expressed in reward-related brain areas. Although most studies have focused on the role of dopamine D1 and D2 receptors in mediating addictive properties of alcohol, D3, D4, and D5 subtypes may also have specific roles in regulating alcohol seeking. However, the lack of selective pharmacological tools has made it difficult to differentiate between receptor subtypes within the D1 and the D2 families.
Extended alcohol intake with periods of withdrawal significantly affects extracellular levels of dopamine (Ericson et al., 2020; Thielen et al., 2004), and alters dopamine D1 and D2 receptor binding sites in brain-subregions such as NAc, dorsal striatum and amygdala (Kim et al., 1997; Sari et al., 2006). Reduced dopamine D2 receptor expression in PFC further parallels with alcohol-induced CPP (Rotter et al., 2012). These changes in dopaminergic neurotransmission may in turn contribute to an imbalance between excitation and inhibition via striatal medium spiny projection neurons (MSNs), which may further promote alcohol seeking (Cheng et al., 2017).
Compounds that increase or stabilize dopamine levels have been shown to prevent reinstatement and suppress relapse-like drinking in the alcohol deprivation effect model [ADE; (Fredriksson et al., 2019; Libarino-Santos et al., 2020; Soderpalm et al., 2020; Spanagel & Holter, 1999; Sutera et al., 2016)]. Inhibition of the dopamine degrading enzyme cathechol-O-methyltransferase (COMT) also reduces cue-induced reinstatement in male rats (McCane et al., 2018). Importantly, both activation and inhibition of dopamine receptor signaling may affect alcohol seeking in a similar manner, either by acting as a replacement treatment analogous to opioid maintenance therapy, or by blocking rewarding effects of alcohol and affecting goal-directed behavior. Alcohol induced changes in dopaminergic neurotransmission appear to be highly time-dependent in relation to alcohol use, complicating interpretation of findings (Hirth et al., 2016).
Dysfunction of D2-like receptor signaling in particular has been associated with alcohol seeking (Blum et al., 1995). Systemic administration or bilateral NAc injection of a dopamine D2 receptor antagonist robustly decreases alcohol seeking responses during extinction trials (Czachowski et al., 2002; Samson & Chappell, 2004), which may be linked to the role of accumbal dopamine D2 receptor for processing information related to stimulus control and goal-directed behavior. After longer exposure periods, the dopamine D2-like receptor dependency appears to shift towards the dorsal striatum (Corbit et al., 2014). Local administration of the combined D1/D2 dopamine receptor antagonist flupenthixol in the dorsolateral striatum (DLS) dose-dependently decreases seeking responses, and the sensitivity to the antagonist predicted vulnerability to subsequent development of compulsive alcohol seeking on a second order schedule (Giuliano et al., 2019). Dorsal striatal dopamine levels are linearly correlated with the persistence of compulsive alcohol seeking, which is seen in a subset of animals (Giuliano et al., 2019). In addition to its effects in the DLS, flupenthixol also suppresses alcohol seeking when administered locally in the amygdala, but not in the NAc core (Gremel & Cunningham, 2009). This is in line with a postulated role of amygdala in the progressive shift from ventral to dorsal striatum as drug seeking behavior becomes an incentive habit (Belin et al., 2009).
The dopamine D3 receptor may be of particular interest when assessing alcohol seeking and cue-induced reinstatement. Cue-induced alcohol-seeking is suppressed by D3 antagonists (Vengeliene et al., 2006). Furthermore, systemic administration of a D3 antagonist, or a partial agonist, suppresses relapse-like behavior following alcohol deprivation in long-term alcohol drinking Wistar rats. Alcohol-induced up-regulation of dopamine D3 receptors in this model is particularly prominent in the dorsal striatum, suggesting that it may contribute to alcohol-seeking and relapse (Vengeliene et al., 2006).
In cocaine self-administration, a seminal paper proposed a model for individual addiction vulnerability, based on three criteria thought to parallel key clinical phenomena of addiction: inability to abstain during a signaled period of reward unavailability, increased motivation assessed using a progressive ratio schedule, and persistent alcohol intake despite aversive foot shocks. In this model, the minority of rats that met all three criteria also showed increased cocaine-seeking, both when induced by priming and by drug-associated cues (Deroche-Gamonet et al., 2004). A similar model was recently applied to alcohol (Domi et al., 2019; Jadhav et al., 2017, 2018). Rats that reached all three criteria showed increased dopamine D1 and decreased dopamine D2 receptor mRNA expression in the DLS three months later. This supports a role for dopaminergic dysregulation in compulsive alcohol seeking and suggests that dopaminergic neuroadaptations may persist even after protracted abstinence. Interestingly, while cue-induced reinstatement is insensitive to D4 antagonists, blockade of D4 receptors suppresses stress-induced reinstatement (Kim et al., 2020).
Taken together, dopamine D1-like receptor signaling in the NAc appears to be important for regulating alcohol intake, while D2-like receptors in the dorsal striatum seem to be most important for alcohol seeking and reinstatement (Table 1). At the same time, dopamine D1 receptors in the dorsal mPFC play a key role in cocaine-induced reinstatement of cocaine seeking (Devoto et al., 2016), and stress-induced activation of VTA dopamine projection to the PFC has been proposed to induce reinstatement of cocaine-seeking behavior via a glutamatergic projection to the NAc core (McFarland et al., 2004). Since ablation of mPFC neurons projecting to the NAc has been shown to block cue-induced reinstatement of alcohol seeking (Keistler et al., 2017), it is possible that similar pathways are also recruited during cue-guided alcohol seeking.
2.4 Section IV: glutamatergic signaling in alcohol seeking behaviors
Alcohol seeking has been especially linked to glutamatergic changes in amygdalo-cortico-striatal circuits (Burnett et al., 2016), and involves both ionotropic (iGluR) and metabotropic (mGluR) glutamate receptors. It has been proposed that, as alcohol use becomes more compulsive, a transition in neural control occurs from metabotropic to ionotropic receptors (Hwa et al., 2017). However, the role of glutamatergic receptors and their subunit composition in alcohol seeking differs depending on localization (Burnett et al., 2016). Potentiation of glutamatergic activity after prolonged heavy drinking results in long-lasting plasticity mainly in corticostriatal synapses that sustain alcohol seeking (Ma et al., 2017; Meinhardt et al., 2013). Impairments in neuroplasticity produced by chronic alcohol exposure in brain areas involved in cognitive processes may dampen behavioral flexibility and promote habitual seeking behaviors (Kroener et al., 2012; Renteria et al., 2018).
The ventral and dorsal striatum are differentially implicated in alcohol addiction. They receive glutamatergic inputs from both amygdalar and cortical projections that innervate MSNs, and interact with dopaminergic inputs to these cells (Lobo & Nestler, 2011). It has been reported that extracellular levels of glutamate are increased in the basolateral amygdala (BLA) and NAc core during cue-induced reinstatement of alcohol seeking (Gass et al., 2011). Furthermore, the mPFC-NAc pathway is necessary for cue-induced reinstatement of alcohol seeking. Selective ablation of glutamatergic mPFC neurons projecting to NAc but not BLA prevented cue-induced reinstatement, without influencing extinction. Reinstatement was also prevented by ablation of amygdalar projections to the NAc (Keistler et al., 2017).
It has been proposed that, similar to other addictive drugs, alcohol seeking becomes a maladaptive habit that relies on a shift from of control from the ventral and dorsomedial striatum (DMS) to the DLS (Belin & Everitt, 2008; Giuliano et al., 2019; Willuhn et al., 2012). While DMS receives glutamatergic inputs from associative cortices, DLS is innervated by sensorimotor cortex and the thalamus (Bolam et al., 2000; Reig & Silberberg, 2014). Potentiation of glutamatergic inputs to the DLS, together with dopamine-related changes, is one of the main mechanisms behind the emergence of habitual alcohol seeking (Barker et al., 2015; Corbit et al., 2014).
Thus, a plethora of glutamatergic neuroadaptations contribute to the emergence of alcohol addiction-like behaviors, including alcohol seeking. In the following, we describe some molecular mechanisms that involve glutamatergic neurotransmission, and have been shown to be important for alcohol seeking in rodent models (Table 3). We focus on the potential for targeting metabotropic and ionotropic glutamate receptors in order to rescue maladaptive changes that occur with chronic use.
TABLE 3 Compounds targeting glutamatergic neurotransmission in alcohol seeking behavior
TABLE 3. Compounds targeting glutamatergic neurotransmission in alcohol seeking behavior2.4.1 Ionotropic glutamate receptors
Glutamate produces its direct effects on neuronal excitability and firing through iGluRs, N-methyl-d-aspartate receptors (NMDAR), α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptors (AMPAR) and kainate receptors that act as ligand-gated ion channels (Traynelis et al., 2010). These receptors mediate fast excitatory neurotransmission and are critically important for synaptic plasticity in brain regions that mediate alcohol seeking and taking (Bell et al., 2016). Chronic alcohol exposure and withdrawal result in increased activity and expression of both NMDARs and AMPARs, resulting in neuroadaptations that reduce behavioral flexibility (Christian et al., 2012; Krystal et al., 2003; Wang et al., 2012). NMDARs have been extensively studied for their involvement in both cue-induced reinstatement of alcohol seeking and compulsive alcohol seeking, i.e. seeking (rather than taking) behavior that continues despite negative consequences.
As noted in Section I, compulsive alcohol seeking has recently been shown to emerge alongside compulsive drinking (Giuliano et al., 2018, 2019). It has previously been shown that alcohol-induced neuroadaptations of accumbal NMDARs promote alcohol taking that is punished with footshock or quinine adulteration (Seif et al., 2013, 2015). Also, punishment-resistant alcohol-seeking in mice increases following a history of alcohol dependence, and is accompanied by increased expression of NMDAR subunits GluN1 and GluN2A in the medial orbitofrontal cortex (OFC) (Radke et al., 2017). A recent study examined molecular pathways involved in habitual alcohol seeking. A random interval schedule of reinforcement was used to promote the emergence of habitual responding (Dickinson et al., 1983), and satiety-induced outcome devaluation was used to test habitual behavior. The GluN2B NMDAR subunit in the OFC, a brain region that projects to the dorsal striatum, was found to mediate habitual alcohol seeking through a mechanism involving mTORC1 signaling (Morisot et al., 2019). These finding are in agreement with previous reports indicating that activation of mTORC1 signaling is a key mechanism behind heavy alcohol use and relapse (Ben Hamida et al., 2019; Laguesse et al., 2017).
Up-regulation of NMDARs GluN2B subunit expression in corticostriatal circuits is critical for promoting reinstatement of alcohol-seeking (Wang et al., 2010). NMDAR antagonists have shown efficacy in blocking both priming-induced reinstatement, and relapse-like behavior after protracted abstinence in the ADE model (Spanagel, 2009; Vengeliene et al., 2005; Wang et al., 2010). By contrast, NMDARs do not seem to play a role in cue-induced reinstatement of alcohol seeking (Bäckström & Hyytiä, 2004; Eisenhardt et al., 2015). Suppressed cue-induced reinstatement of alcohol seeking was seen with the clinically approved medication acamprosate, and was attributed to NMDA-mediated effects (Bachteler et al., 2005), but it has since become clear that effects of acamprosate are complex, and not likely to be mediated through direct actions at NMDARs (Spanagel et al., 2014).
Similar to NMDARs, AMPAR function is also enhanced after chronic alcohol exposure, and NMDAR-dependent increase in AMPAR activity has been shown to trigger drug seeking (Christian et al., 2012; Gipson et al., 2013; Shen et al., 2011). Accordingly, the AMPA positive allosteric modulator (PAM) aniracetam potentiates cue-induced reinstatement of alcohol seeking in alcohol-preferring P rats (Cannady et al., 2013). Chronic alcohol also disrupts Ca2+ /calmodulin-dependent protein kinase II (CaMKII)-AMPA signaling in the PFC and amygdala, increasing the risk of relapse to alcohol seeking through CaMKII-dependent activation of AMPARs (Cannady, Fisher, et al., 2017; Salling et al., 2017). In agreement with these findings, the selective AMPAR receptor antagonist GYKI 52,466 blocks cue-induced reinstatement and ADE in rats (Sanchis-Segura et al., 2006). Moreover, AMPAR/kainate mixed antagonists (CNQX and NBQX) are able to reduce cue-induced reinstatement of alcohol seeking (Bäckström & Hyytiä, 2004; Czachowski et al., 2012; Sciascia et al., 2015). Selective kainate receptor antagonists such as LY466195 have mostly been studied for their ability to reduce alcohol intake (Van Nest et al., 2017), and their potential to influence alcohol seeking has to our knowledge not yet been studied.
2.4.2 Metabotropic glutamate receptors
A more fruitful category of potential therapeutic targets may be offered by mGluRs, which are widely expressed in both neurons and glial cells of the central nervous system in (Niswender & Conn, 2010)). mGluRs are GPCRs that mediate slow neurotransmission through modulation of second messenger levels and ion-channel activity (Conn & Pin, 1997). They are located in the proximity of the synaptic cleft in both pre-and postsynaptic neurons (Cartmell & Schoepp, 2000; Shigemoto et al., 1997). Briefly, eight metabotropic glutamate receptors have been identified, and categorized into three groups, based on sequence similarity, signal transduction pathways, and pharmacological properties. Group I consists of mGluR1 and 5; group II of mGluR2 and 3; while mGluR4, 6, 7, and 8 belong to Group III. Group I mGluRs are Gq-coupled, are mainly present in postsynaptic neurons, and when activated, trigger a cascade that ultimately results in increased intracellular Ca2+ levels. Activation of Group I mGluRs also triggers changes in transcriptional regulation and gene expression. Group II and III, which are Gi/o coupled, are mostly localized at presynaptic terminals and in astrocytes, where they modulate the release of glutamate and other neurotransmitters (Wang & Zhuo, 2012; Yin & Niswender, 2014). The involvement of mGluRs in alcohol seeking has been extensively studied, with most findings focusing on mGluR5 and 2, which have been considered potential medication targets.
Alcohol acutely dampens mGluR1/5 function, but protracted alcohol use potentiates both the expression and activity of these receptors (Zorumski et al., 2014). Using a drug discrimination procedure, it was shown that activation of accumbal mGluR5s is essential for interoceptive effects of alcohol (Besheer et al., 2009). Accordingly, competitive mGluR5 antagonists as well as mGluR5 negative allosteric modulators (NAMs) attenuate cue-induced reinstatement of alcohol seeking, both when administered systemically, and when microinjected into the NAc or the BLA (Bäckström et al., 2004; Caprioli et al., 2018; Sinclair et al., 2012). Using the selective mGluR5 NAM 2-Methyl-6-(phenylethynyl)pyridine (MPEP), it was shown that suppressed reinstatement of alcohol seeking following down-regulated mGluR5 transmission involves the extracellular signal-regulated kinases 1/2 (ERK1/2) signaling pathway (Schroeder et al., 2008). ERK1/2 signaling is downstream of mGluR5, and is activated in amygdala inputs to the ventral striatum by contingent presentation of alcohol associated cues. Within this circuitry, ERK1/2 phosphorylation was associated with increased cue-induced reinstatement of alcohol seeking, and this effect was counteracted by MPEP (Schroeder et al., 2008).
A potential interpretation of these findings is that mGluR5s are involved in associative learning that links alcohol-associated cues with alcohol effects, becomes progressively strengthened over the course of developing alcohol addiction, persists into protracted abstinence, and contributes to alcohol seeking under non-reinforced conditions. In addition to blocking the recall of these alcohol-memories as reviewed above, facilitating their extinction may also offer treatment opportunities. Exposure-based extinction of alcohol cue-reactivity is a clinical treatment of alcohol addiction, but its efficacy is limited (Mellentin et al., 2017), and could potentially be strengthened using medications. In that context, the mGluR5 PAM CDPPB (3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide) has been shown to facilitate extinction of cue-conditioned alcohol seeking (Gass et al., 2014). This effect was mediated through mGluR5 modulation of small-conductance calcium activated potassium (KCa2) channels, and was obtained both with systemic and intra-infralimbic/PFC activation of mGluR5s (Cannady, McGonigal, et al., 2017).
In contrast with mGluR5, mGluR1 effects on alcohol seeking have not been extensively studied, with most of the literature focusing on mGluR1 PAMs and NAMs effects on alcohol consumption (Besheer et al., 2008; Cozzoli et al., 2014; Lum et al., 2014).
mGluR2-mediated control of glutamatergic neurotransmission through presynaptic modulation of glutamate release has received considerable interest as a pharmacological target in several psychiatric disorders (Crupi et al., 2019). Prolonged alcohol exposure has been shown to disrupt mGluR2 function by down-regulating expression of Grm2, the gene encoding this receptor. Deficits in corticostriatal and cortico-amygdala mGluR2-mediated feedback inhibition of glutamate release have been shown to promote reinstatement of alcohol seeking (Lovinger & McCool, 1995; Meinhardt et al., 2013). High levels of glutamate in the BLA and NAc have been detected during cue-induced reinstatement of alcohol seeking, together with an alcohol-induced mGluR2 down-regulation in mPFC (Gass et al., 2011; Meinhardt et al., 2013). Genetically selected alcohol-preferring P rats lack mGluR2s, and show escalation of alcohol consumption and resistance to alcohol drinking devaluation (Timme et al., 2020; Zhou et al., 2013).
In agreement with the findings reviewed above, the mixed mGluR2/3 agonist LY379268 has been shown to reduce reinstatement of alcohol seeking, both when reinstatement was induced by cues and by footshock stress (Bäckström & Hyytiä, 2005; Kufahl et al., 2011; Sidhpura et al., 2010; Zhao et al., 2006). However, LY379268 and other orthosteric group II mGluR agonists are unable to discriminate between the respective contribution of mGluR2 and mGluR3. In a recent paper, we therefore tested the effect of the selective mGluR2 PAM AZD8529 on alcohol taking and seeking behaviors (Augier et al., 2016). Cue- but not stress-induced reinstatement of alcohol seeking was potently blocked by AZD8529 in Wistar rats. This effect was absent in alcohol preferring P rats lacking functional mGluR2s. Of note, AZD8529 also reduced 20% alcohol self-administration, but this effect was marginal compared to the potent and specific blockade of reinstatement induced by alcohol-associated stimuli. Together with previous findings obtained with mixed mGluR2/3 agonists, this indicates that mGluR2 are specifically involved in cue-induced alcohol seeking. A potential role of mGluR3 remains to be evaluated. Thus, mGluR2 agonists or PAMs may have therapeutic potential, and prevent relapse to alcohol use.
Results are generally less promising with manipulations targeting group III mGluRS, mGluR7, and mGluR8. Upon intracerebroventricular administration, a mixed mGlu4/mGlu7 agonist was reported to reduce reacquisition of alcohol self-administration after a period of abstinence (Lebourgeois et al., 2018); while the GluR8 receptor agonist (S)-3,4-DCPG attenuated cue-induced reinstatement of alcohol seeking, but only at doses with motor-suppressant effects (Bäckström & Hyytiä, 2005).
2.4.3 Targeting glutamatergic transmission for therapeutic purposes
The findings reviewed here clearly support the notion that glutamatergic transmission plays a critical role in alcohol seeking and relapse. The complexity of ionotropic glutamate receptors and their subunits may seem to offer a rich landscape for development of therapeutics for alcohol addiction. Decades of experience from attempts to develop medications targeting NMDA receptors for ischemic excitotoxicity, epilepsy and Parkinson's disease, to name a few indications that have been pursued, suggest otherwise (Low & Roland, 2004). One possible conclusion is that ionotropic effects of glutamate may simply be too fundamental for brain function to allow modulation for therapeutic purposes while maintaining adequate safety and tolerability. In contrast, preclinical findings indicate that presynaptic agonists of mGluR2/3 receptors and antagonists of postsynaptic mGluR5 may be effective pharmacotherapeutic treatments in alcohol seeking behavior and relapse.
2.5 Section V: GABAergic neurotransmission and alcohol seeking
GABAergic neurotransmission plays an important role in a wide range of alcohol effects, from initial intoxication to alcohol seeking and relapse (Enoch, 2008; Heilig et al., 2011). GABA is the principal inhibitory neurotransmitter of the brain, and inhibits the activity of mesolimbic dopamine neurons (Bowery & Smart, 2006). It acts on two classes of receptors: ionotropic GABAA receptors, which are ligand-gated chloride channels that also include the GABAA ρ subclass (previously known as GABAC receptor), and the metabotropic GABAB receptor, that is G protein coupled, and regulates the activity of potassium and calcium channels (Bormann, 2000; Kuriyama et al., 1993). Alcohol affects GABAergic transmission both via pre- and post-synaptic mechanisms, and has both acute and long-term effects on GABAergic transmission (Roberto & Varodayan, 2017). Increased propensity for relapse, driven by negative reinforcement, has been suggested to result from a dysregulation of peptidergic neuromodulatory systems that converge on GABAergic circuitry within the extended amygdala (George & Hope, 2017; Koob, 2008). The extended amygdala comprises the CeA, the NAc shell, and the lateral portion of the BNST (Heimer & Alheid, 1991).
CeA plays a role in both footshock- and yohimbine-induced alcohol seeking (Walker et al., 2017, 2020). In vivo studies and ex vivo slice electrophysiology studies show that chronic alcohol exposure, which promotes a range of alcohol addiction-like behaviors, increases GABA release in the CeA through both pre- and post-synaptic mechanisms (Roberto et al., 2003, 2004). In agreement with those observations, we recently found that impaired clearance of GABA within the CeA, caused by a low expression of the GABA transporter GAT-3, is associated with pathological high choice for alcohol over a natural reward, and continued self-administration of alcohol despite punishment and adulteration with quinine. Decreased expression of GAT-3 in the amygdala was accompanied by decreased expression of several GABAA receptor subunit transcripts, presumably reflecting a compensatory down-regulation in response to the sustained increase in GABAergic tone (Augier et al., 2018). Of note, SP-mediated activation of NK1 receptors in CeA promotes GABAergic transmission in this structure, and this effect is potentiated in rats with a history of alcohol dependence (Khom et al., 2020). These finding suggest that pharmacological interventions with an ability to restore GABA homeostasis within the CeA may have therapeutic potential in alcohol addiction (Heilig et al., 2019; Spanagel, 2018).
GABAergic transmission in the BNST also contributes to stress-induced reinstatement of drug seeking and negative affect associated with addiction (Lebow & Chen, 2016; Lowery-Gionta & Kash, 2014). BNST is a major target of CeA projection neurons, about 80% of which are GABAergic (Gungor & Pare, 2016; Le Gal LaSalle et al., 1978; Weller & Smith, 1982). Pina and colleagues have shown that electrolytic ablation of the BNST both before and after conditioning reduced the magnitude of cue-induced alcohol seeking in mice (Pina et al., 2015).
BNST sends extensive projections to the VTA (Dong & Swanson, 2004, 2006; Kudo et al., 2012). These predominantly innervate non-dopaminergic VTA neurons, and are made up by distinct populations whose activity promotes divergent motivational states. Specifically, a majority of VTA-projecting BNST neurons are GABAergic, and their activation produces anxiolytic-like and rewarding effects. In contrast, activity of a smaller population of glutamatergic BNST inputs to the VTA is anxiogenic and aversive (Jennings et al., 2013; Kudo et al., 2012). A population of CRH-expressing GABA neurons intrinsic to the BNST controls the balance between these BNST outputs by inhibiting the activity of the anxiolytic output neurons, and is itself under influence of serotonergic inputs from the dorsal raphe nucleus (Marcinkiewcz et al., 2016). A recent report showed that chronic intermittent alcohol exposure results in dysregulation of this local GABAergic BNST microcircuit, an effect which may promote negatively reinforced alcohol seeking during withdrawal and protracted abstinence (Pati et al., 2020). In this study, withdrawal from alcohol exposure resulted in increased excitability of the CRH-expressing GABAergic interneurons, accompanied by decreased activity of putatively anxiolytic-acting non-CRH BNST neurons that project to both lateral hypothalamus (LH) and VTA.
In the striatum, chronic alcohol exposure reduces GABAergic transmission, and this effect may separately contribute to increased alcohol seeking and intake (Lovinger & Kash, 2015). After prolonged alcohol exposure, a decrease in GABAergic transmission has been shown in the DLS, DMS and NAc of both mice and monkeys (Cuzon Carlson et al., 2011; Liang et al., 2014; Wilcox et al., 2014). This is accompanied by decreases in both the amplitude and the frequency of miniature inhibitory postsynaptic currents (mIPSCs), suggesting that the underlying mechanism may be either a decrease of GABA release from existing synapses, or a decrease in the number of GABAergic synapses onto dorso-striatal MSNs (Lovinger & Kash, 2015). Alcohol-mediated disinhibition of the DLS may thus result from weakened GABAergic inhibition of this structure, providing a potential mechanistic basis for habitual alcohol seeking (Corbit et al., 2012).
Alcohol-induced changes in GABAergic transmission are in part attributable to changes in the function and sensitivity of GABAA and GABAB receptors (Davies, 2003; Gass & Olive, 2012; Grobin et al., 1998; Kumar et al., 2009). This makes it important to understand adaptations of GABAergic receptors in the different stages of alcohol addiction.
2.5.1 GABAA receptors in relapse to alcohol seeking
GABAA and GABAB receptors both contribute to acute as well as chronic effects of alcohol, including sedation, tolerance, withdrawal, and motivational effects (Chester & Cunningham, 2002; Colombo et al., 2004; June et al., 2003; Lobo & Harris, 2008; Olsen & Liang, 2017).
GABAA receptors are ligand-gated chloride channels composed of five subunits arranged around a central pore. They are assembled from α(1–6) and β(1–3) subunits which are obligate. Assembled receptors may also contain γ(1–3), δ, ε, π, or θ subunits. In addition, ρ(1–3) subunits exist, but do not co-assemble with classical GABAA receptors; instead, they homooligomerize to form GABAA ρ receptors. The composition from multiple subunits allows for an extensive heterogeneity of receptor formation, varies between brain regions, and determines the pharmacological profile of the receptor, including its responses to alcohol (Olsen & Sieghart, 2009). Acute and chronic alcohol exposure induces transient changes in GABAA receptor subunit levels, composition, and regional and subcellular localization (Kumar et al., 2009).
Alcohol potentiates GABAA receptor-mediated synaptic transmission via an increase in GABA release from presynaptic terminals in a multitude of brain regions (Spanagel, 2009), also including the cerebellar cortex (Valenzuela & Jotty, 2015). Prolonged exposure to chronic alcohol facilitates GABAergic transmission, and the rebound renders the brain hyperexcitable during alcohol withdrawal (Lovinger, 2008; Roberto et al., 2012; Steffensen et al., 2009). Chronic alcohol exposure can induce GABAA receptor down-regulation, producing tolerance and withdrawal, and disrupting a wide range of behaviors (Forstera et al., 2016). GABAA receptors in the CeA regulate alcohol-maintained responding in alcohol preferring rats (Foster et al., 2004), but there is little evidence for a role of GABAA receptors in alcohol seeking behaviors.
A recent study showed that chronic administration of the GABAA α1-preferring antagonist, 3-isopropoxy-β-carboline hydrochloride (5–20 mg/kg), selectively reduced alcohol self-administration but not alcohol seeking assessed under a chained schedule of reinforcement in baboons (Holtyn et al., 2017). By contrast, the same compound failed to attenuate drinking in rhesus macaques (Sawyer et al., 2014), while in alcohol preferring P rats, the analogue 3- β-carboline hydrochloride reduced alcohol maintained responding when injected in the ventral pallidum, a key node in neural circuits that control relapse to alcohol seeking (Harvey et al., 2002; Prasad & McNally, 2020). In mice lacking the GABAA α1 receptor subunit, reduction of alcohol drinking was also accompanied by a reduction in saccharin and sucrose consumption (Blednov et al., 2003; June et al., 2007), suggesting that these effects are not specific for alcohol.
2.5.2 Role of GABAB receptors in alcohol seeking
In contrast with the limited and contradictory evidence on the role of GABAA receptors in alcohol seeking, extensive data have accumulated over recent years for a role of GABAB receptors in this behavior (see Table 4).
TABLE 4. Compounds targeting GABAergic neurotransmission in alcohol seeking behavior
GABAB receptors are G-protein coupled, and require dimerization of two subunits (GABAB1 and GABAB2) to be functional (Robbins et al., 2001). Both pre-and postsynaptic activation of GABAB receptors inhibit neurotransmitter release through neuronal hyperpolarization, which results from increased potassium and decreased calcium permeability (Bettler & Tiao, 2006). Preclinical and clinical evidence suggests that activation of GABAB receptors holds promise as a mechanism for treatment of alcohol addiction (Addolorato et al., 2009; Augier, 2021; Farokhnia et al., 2018; Heilig & Egli, 2006).
Results with GABAB activation in animal models of alcohol addiction were recently reviewed (Holtyn & Weerts, 2020). In brief, much of the data have been obtained with baclofen, a selective GABAB receptor agonist that for more than four decades has been clinically approved as a treatment for muscle spasticity. Baclofen has been shown to reduce the reinforcing properties of alcohol and the severity of alcohol withdrawal. It also prevents reinstatement of alcohol seeking [recently reviewed in (Colombo & Gessa, 2018)]. In the alcohol deprivation model of relapse-like drinking, baclofen (1, 1.7 and 3 mg/kg, i.p.) abolished alcohol intake of Sardinian alcohol preferring (sP) rats after 7 days of abstinence (Colombo et al., 2003). These findings were then replicated with baclofen (3 mg/kg i.p.) in sP rats using different alcohol concentrations (10%, 20%, and 30% (v/v) (Colombo et al., 2006). However, following long-term voluntary alcohol drinking with repeated deprivation cycles, chronic baclofen (1 and 3 mg/kg i.p.) reduced relapse-like drinking only at the highest dose. This effect was not specific, since this dose also induced sedative effects and altered food intake inducing a significant body loss (Vengeliene et al., 2018).
Thus, in rodent models, the separation between baclofen doses with specific effects on alcohol-related behaviors and doses that produce non-specific sedative effects or otherwise impair performance (i.e. the therapeutic index) is limited, and variable. The variable findings on baclofen doses required in different relapse studies may be explained by the use of different strains and lines of rats, because of innate differences in their GABAergic transmission. For instance, genetically selected msP rats show increased GABA levels in the CeA at baseline compared to Sprague Dawley and Wistar rats, and this may render them more sensitive to inhibition of GABA release by GABAB activation (Herman et al., 2013). Two additional factors that may influence the dose–response relationship of baclofen are differences in alcohol drinking history, and the acute versus chronic nature of baclofen administration.
Cue-induced relapse to alcohol seeking is reduced by baclofen across species, including rats (both msP and Wistar), as well as baboons, a species in which baclofen also facilitated extinction of responding for alcohol (Duke et al., 2014; Maccioni et al., 2008; Vengeliene et al., 2018). Baclofen has also been reported to reduce yohimbine-induced relapse to alcohol seeking in rats, and to attenuate alcohol seeking in an odor recognition task in mice, in the latter model presumably by blunting the corticosterone response to the footshock (Rabat et al., 2019; Williams et al., 2016). The latter effect is in line with a well-documented clinical relationship between HPA axis activity, craving, and relapse to alcohol use (Blaine & Sinha, 2017; Sinha et al., 2011; Stephens & Wand, 2012). It is also in agreement with a recent clinical study that reported reduced cortisol levels in alcohol-dependent patients that received treatment with baclofen (Geisel et al., 2019).
Clinically, baclofen has shown promising, although conflicting, results for the treatment of alcohol addiction (Addolorato et al., 2007; Pierce et al., 2018; Agabio et al., 2018; see also Agabio & Leggio, 2018; Augier, 2021 for review on this topic), but safety and tolerability concerns have prevented its approval as a clinical alcohol addiction treatment (ANSM, 2017; Bowery, 2006; Garbutt, 2018). Some of these concerns are related to tolerance and dose-escalation, phenomena expected with chronic GPCR agonist treatment. PAMs of the GABAB receptor have the potential to avoid these effects, by targeting a site that is topographically distinct from the orthosteric GABA binding sites, and instead potentiating the effect of GABA upon its binding to the receptor (Froestl, 2010; Perdona et al., 2011). GABAB PAMs have received considerable interest as potential therapeutics for alcohol addiction in recent years (Augier, 2021; Holtyn & Weerts, 2020; Maccioni & Colombo, 2019). The majority of studies that have examined GABAB PAMs support their improved selectively and wider therapeutic index in reducing alcohol seeking and taking.
For instance, we have recently found that the selective GABAB PAM, ADX71441 (3, 10 mg/kg), potently suppressed both cue- and stress (footshock)-induced reinstatement of alcohol seeking in Wistar rats (Augier, Dulman, Damadzic, et al., 2017). Moreover, ADX71441 attenuated stress-induced neuronal activity, indexed by cFos activity, in an interconnected network of brain structures that included NAc shell, mPFC and the dorsal raphe nucleus. Surprisingly, although neuronal activity in the CeA was also reduced by ADX71441 (3 mg/kg), it did not correlate with relapse-like behavior. Based on these findings, ADX71441 may act on distinct but converging networks that mediate relapse, or, alternatively, on a common neuronal pathway that promotes alcohol seeking initiated by stress, drug cues or drug priming (Kalivas & Volkow, 2005).
In addition, the novel GABAB PAM, CMPPE, (10, 30 mg/kg i.p), reduced relapse to alcohol drinking in a repeated alcohol deprivation model, and suppressed cue-induced reinstatement of alcohol seeking in Wistar rats (Vengeliene et al., 2018). It also showed efficacy in reducing cue-induced reinstatement of alcohol seeking when administered at lower doses (2.5, 5 mg/kg i.p) in alcohol preferring sP rats (Maccioni, Fara, et al., 2019). Another novel GABAB PAM, COR659, suppressed cue-induced reinstatement of alcohol seeking when administered at the lowest dose tested (2.5 mg/kg), and reduced alcohol self-administration in a manner that was maintained when the compound was administered chronically (Maccioni, Colombo, et al., 2019).
The mechanism(s) through which GABAB activation prevents relapse to alcohol seeking remain unclear. One candidate mechanism is through inhibition of mesolimbic dopaminergic neurons. Activation of GABAB receptors in the VTA results in a decreased dopamine release both in the NAc and the mPFC. Moreover, a GABAB PAM injected in the VTA enhanced GABAB inhibition of dopaminergic neuronal firing (Chen et al., 2005). Presumably related to these effects, microinjections of baclofen into the VTA dose-dependently suppressed cue-induced alcohol seeking in rats (Leite-Morris et al., 2008). It was recently reported that dissociable mesolimbic dopamine pathways control responding triggered by discrete alcohol-associated cues and alcohol associated contexts, respectively. Inhibition of inputs from the VTA to the NAc core reduced alcohol seeking triggered by an alcohol associated cue, irrespective of context. In contrast, silencing terminals of VTA inputs to the NAc shell selectively reduced cue-induced alcohol seeking in an alcohol-associated context (Valyear et al., 2020). If activation of GABAB receptors can attenuate activity of VTA neurons that belong to both these populations, it may therefore be able to broadly prevent relapse both when induced by discrete cues, and by contextual stimuli.
In summary, activation of GABAB receptors appears to hold considerable promise as a treatment for prevention of alcohol seeking and relapse. GABAB PAMs may be able to avoid the safety and tolerability issues that limit the use of the existing orthosteric GABAB agonist baclofen. Developing GABAB PAMs that are safe for human use, and evaluating their efficacy as alcohol addiction medications is currently one of the most promising avenues for bringing forward novel therapeutics for this indication.
3 CONCLUDING REMARKS
Since their introduction in the early 1980s, animal models that use reinstatement of drug seeking following extinction have been widely used to study mechanisms of relapse. The application of these models to alcohol seeking has generated a vast literature, most (but not all) of which we have reviewed here. This work has made major advances in the past decades, and has identified multiple biological systems that contribute to alcohol seeking.
The overarching question ahead of the field is whether these advances and the biological systems they have identified are able to predict activity to prevent craving and relapse in people with alcohol addiction. Because of multiple failures in clinical development, industry efforts to develop psychiatric medications have dramatically decreased, and the ability of animal models to predict clinical activity in psychiatric disorders in general has been broadly questioned (Hyman, 2014). The landscape in addictive disorders is no different (Venniro et al., 2020).
So, are studies of alcohol seeking in animals a worthwhile effort? Some insights into this critical question may be gained from the present review. Specifically, we do not believe that data support any blanket conclusions that animal models of alcohol-seeking and relapse are, or are not, able to predict clinical activity in a meaningful way. The landscape that has emerged is in fact much more complex. On one hand, human and animal findings obtained with naltrexone and other tools targeting MOP strongly support a predictive validity of both priming- and cue-induced reinstatement models. Although somewhat more complex, findings with baclofen and GABAB activation also generally support a predictive validity of relapse models in animals. On the other hand, the lack of success with CRH1 antagonists in clinical development, together with other data, may seem to prompt just the opposite conclusion.
Our preliminary attempt to integrate these observations is that a more nuanced view of the animal findings is needed. Predictive validity is not an all or nothing phenomenon, and is not necessarily first and foremost a property of the model. Instead, it is likely to vary dramatically across biological systems studied. A key factor that determines this variation is probably the degree to which the respective system and its elements are conserved across species, something that has received far too little attention. One implication of this realization is that dedicating decades of preclinical studies to any particular biological system may not be the optimal strategy, at least not if neuroscience of alcohol addiction wants to contribute to better outcomes for patients. Instead, signals from animal models should as quickly as possible be put to test in human proof-of-principle, biomarker-based studies, as outlined and recently tested by the NIMH Fast-Fail initiative for KOP antagonism in depression (Pizzagalli et al., 2020). If human translation is not supported, continued preclinical work is associated with a high opportunity cost.
With this type of strategy in mind, we believe that a priority list of targets for clinical fast-fail evaluation is suggested by the literature reviewed here. At the top of this list, in our assessment, is development of safe and well-tolerated GABAB PAMs, which should be evaluated for prevention of both stress- and cue-induced craving and relapse in patients. A priority that comes in a close second is evaluation of KOP antagonists, which based on the data reviewed here would be predicted to prevent stress-induced craving and relapse, potentially offering a useful combination with naltrexone. In third position is evaluation of mGluR2 PAMs. We are less optimistic about interventions targeting dopaminergic or ionotropic glutamate receptors, in part because of the fundamental role these have on brain function and behavior, which may make it difficult to develop medications that combine efficacy with acceptable safety and tolerability.