Abstract
Drug addiction represents a dramatic dysregulation of motivational circuits that is caused by a combination of exaggerated incentive salience and habit formation, reward deficits and stress surfeits, and compromised executive function in three stages. The rewarding effects of drugs of abuse, development of incentive salience, and development of drug-seeking habits in the binge/intoxication stage involve changes in dopamine and opioid peptides in the basal ganglia. The increases in negative emotional states and dysphoric and stress-like responses in the withdrawal/negative affect stage involve decreases in the function of the dopamine component of the reward system and recruitment of brain stress neurotransmitters, such as corticotropin-releasing factor and dynorphin, in the neurocircuitry of the extended amygdala. The craving and deficits in executive function in the so-called preoccupation/anticipation stage involve the dysregulation of key afferent projections from the prefrontal cortex and insula, including glutamate, to the basal ganglia and extended amygdala. Molecular genetic studies have identified transduction and transcription factors that act in neurocircuitry associated with the development and maintenance of addiction that might mediate initial vulnerability, maintenance, and relapse associated with addiction.
Conceptual framework, definitions, and animal models
Drug addiction can be defined as a chronically relapsing disorder, characterised by compulsion to seek and take the drug, loss of control in limiting intake, and emergence of a negative emotional state (eg, dysphoria, anxiety, irritability) when access to the drug is prevented. From a diagnostic perspective, the term addiction is now encompassed by the term substance use disorders. In 2013, DSM-51 combined what was previously conceptualised as two separate and hierarchical disorders (substance abuse and substance dependence) into one construct, defining substance use disorders on a range from mild to moderate to severe, with the severity of an addiction depending on how many of the established criteria apply.
Although much of the early research with animal models focused on the acute rewarding effects of drugs of abuse, focus is shifting to the study of chronic drug administration-induced brain changes that decrease the threshold for relapse, which corresponds more closely to human imaging studies of individuals who have substance use disorders. One of the main goals of neurobiological research is to understand changes at the molecular, cellular, and neurocircuitry levels that mediate the transition from occasional, controlled substance use to loss of control in drug intake and chronic addiction. Because only some substance users make this transition, neurobiological factors that influence the diverse individual differences in drug responses have also attracted increasing interest. Cogent arguments have been made that addictions are similar to other chronic relapsing disorders—with individual differences in responses to the same exogenous challenges and limited efficacy of treatment—such as diabetes, asthma, and hypertension.
The purpose of this Review is twofold. First, we aim to elaborate a heuristic framework based on the neuropsychopharmacological and brain imaging phenotype of addiction in the context of three functional domains (incentive salience, negative emotional states, and executive function) mediated by three major neuro-biological circuits (basal ganglia, extended amygdala, and prefrontal cortex). Second, we aim to identify neurochemically defined mini circuits that can independently or interactively mediate functional neuroplasticity within the three major circuits to produce incentive salience and compulsive-like habits, negative emotional states of low reward and excessive stress, and compromised executive function.
Building on previous identification of the three overall domains, this Review provides a framework for integration of the ever-expanding neuroplastic complexity of motivational systems that are involved in addiction and for identification of new targets for diagnosis, treatment, and prevention of addiction. Addiction can be conceptualised as a three-stage, recurring cycle—binge/intoxication, withdrawal/negative affect, and preoccupation/anticipation (craving)—that worsens over time and involves neuroplastic changes in the brain reward, stress, and executive function systems. Derived from a confluence of information from social psychology of human self-regulation failure, psychiatry, and brain imaging, these three stages provide a heuristic framework for the study of the neurobiology of addiction.
A definition of impulsivity is “a predisposition toward rapid, unplanned reactions to internal and external stimuli without regard for the negative consequences of these reactions to themselves or others.” A definition of compulsivity is the manifestation of “perseverative, repetitive actions that are excessive and inappropriate.” Impulsive behaviours are often accompanied by feelings of pleasure or gratification, but compulsions in disorders such as obsessive-compulsive disorder are often performed to reduce tension or anxiety from obsessive thoughts. In this context, individuals move from impulsivity to compulsivity, and the drive for drug-taking behaviour is paralleled by shifts from positive to negative reinforcement (figure 1). However, impulsivity and compulsivity can coexist, and frequently do so in the different stages of the addiction cycle.
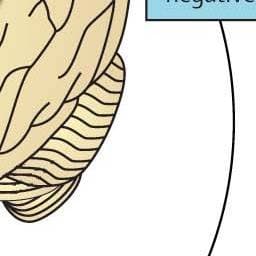
Figure 1: Model of interacting circuits in which disruptions contribute to compulsive-like behaviours underlying drug addiction. The overall neurocircuitry domains correspond to three functional domains: binge/intoxication (reward and incentive salience: basal ganglia [blue]), withdrawal/negative affect (negative emotional states and stress: extended amygdala and habenula [red]), and preoccupation/anticipation (craving, impulsivity, and executive function: PFC, insula, and allocortex [green]). Arrows depict major circuit connections between domains, and numbers refer to neurochemical and neurocircuit-specific pathways known to support brain changes that contribute to the allostatic state of addiction. PFC=prefrontal cortex. ACC=anterior cingulate cortex. OFC=orbitofrontal cortex. NAc-VTA=nucleus accumbens-ventral tegmental area. Modified from Koob and Volkow (2010) with permission from Nature Publishing Group.
Understanding of the neurobiology of addiction has progressed through the study of animal models and, more recently, through brain imaging studies in individuals with addiction. Although no animal model of addiction fully emulates the human condition, they permit investigations of specific signs or symptoms that are associated with the psychopathological condition. If the model adequately mimics the phenomenology observed in humans as they transition from experimentation to addiction, then it is more likely to have construct or predictive validity. The phenomena under study can be models of different systems (genetic, epigenetic, transcription, cellular, and network), psychological constructs (positive and negative reinforcement), symptoms outlined by psychiatric nosology (craving, hypohedonia, and dysphoria), and stages of the addiction cycle.
Recently developed animal models take advantage of individual and strain diversity in responses to drugs, incorporate complex environments with access to and choices of alternative reinforcers, and test effects of stressful stimuli, allowing the investigation of neuro biological processes that underlie the risk for addiction and environmental factors that provide resilience against vulnerability. Animal models have also started to explore the influence of developmental stage and sex in drug response to better understand the greater vulnerability to substance use disorders when drug use is initiated in adolescence, and the distinct drug use trajectories that are observed in men and women. The neurobiological mechanisms involved in the stages of the addiction cycle can be conceptualised as domains, with a focus on specific brain circuits, the molecular and neurochemical changes in those circuits during the transition from drug taking to addiction, and the way in which those changes persist in the vulnerability to relapse. Equally convincing, animal models of the specific stages of the addiction cycle can be paralleled by human laboratory models and studied with neuroimaging.
Neurobiological mechanisms of the binge/intoxication stage
Drug reward
Drugs of abuse activate brain reward systems, and research on drug addiction has in large part defined the neurocircuitry of reward. This line of investigation is fundamental because changes to how the drug-induced reward system is activated are key to the understanding of the development of addiction. Reward is defined herein as any event that increases the probability of a response with a positive hedonic component. A principal focus of research on the neurobiology of the rewarding effects of abused drugs has been the origins and terminal areas of the ascending mesocorticostriatal dopamine systems that have a key role in the rewarding properties of nearly all drugs of abuse. In humans, positron emission tomography studies have shown that intoxicating doses of alcohol and drugs release dopamine and opioid peptides into the ventral striatum, and that fast and steep release of dopamine is associated with the subjective sensation of the so-called high. This is because fast and steep increases in dopamine activate low-affinity dopamine D1 receptors, which are necessary for the rewarding effects of drugs and for triggering conditioned responses. By contrast, dopamine stimulation of high-affinity dopamine D2 receptors is not sufficient for drug reward, and these receptors might even limit drug reward. In this respect, drugs emulate the increases in dopamine triggered by phasic dopamine firing, which are the firing frequencies of dopamine neurons associated with rewarding stimuli42.
Table 1:
Neurotransmitter systems involved in the neurocircuitry of addiction stages and functional domains
Response | |
Binge/intoxication | |
Dopamine | Increase |
Opioid peptides | Increase |
Serotonin | Increase |
γ-aminobutyric acid | Increase |
Acetylcholine | Increase |
Withdrawal/negative affect | |
Corticotropin-releasing factor | Increase |
Dynorphin | Increase |
Norepinephrine | Increase |
Hypocretin (orexin) | Increase |
Substance P | Increase |
Dopamine | Decrease |
Serotonin | Decrease |
Opioid peptide receptors | Decrease |
Neuropeptide Y | Decrease |
Nociceptin | Decrease |
Endocannabinoids | Decrease |
Oxytocin | Decrease |
Preoccupation/anticipation | |
Dopamine | Increase |
Glutamate | Increase |
Hypocretin (orexin) | Increase |
Serotonin | Increase |
Corticotropin-releasing factor | Increase |
The specific circuitry that is associated with drug reward has been broadened to include many neural inputs and outputs that interact with the basal forebrain. As the understanding of the relevant circuits has evolved, so too has the understanding of the relevant neurotransmitters and neuromodulators, which include not only dopamine and opioid peptides but also γ-aminobutyric acid (GABA), glutamate, serotonin, acetylcholine, and endocannabinoid systems that act at the level of either the ventral tegmental area or nucleus accumbens (figure (figure1;1; tables tables1,1, ,22 for neurotransmitter systems and specific neurocircuits involved). Balanced circuits result in proper inhibitory control and decision making and normal functioning of reward, motivation, stress, and memory circuits. These circuits also interact with circuits that are involved in mood regulation, including stress reactivity (which involves the amygdala, hypothalamus, and habenula) and interoception (which involves the insula and anterior cingulate cortex and contributes to the awareness of negative emotional states). Drugs of abuse usurp executive function circuits, motivational circuits, and stress circuits via multiple neurotransmitter-specific neuroplasticity circuits (tables (tables1,1, ,2).2). Key neurotransmitters that are implicated in these neuroadaptations include dopamine, enkephalins, glutamate, γ-aminobutyric acid, norepinephrine, corticotropin-releasing factor (CRF), dynorphin, neuropeptide Y, and endocannabinoids (tables (tables1,1, ,22).
Table 2:
Molecular neurocircuits as focal points for neuroplasticity in addiction
Neurotransmitter | |
Binge/intoxication | |
Ventral tegmental area (circuit 1) | Glutamate |
Ventral tegmental area (circuit 2) | γ-aminobutyric acid |
Dorsal striatum (circuit 3) | Dopamine |
Dorsal striatum (circuit 4) | Glutamate |
Withdrawal/negative affect | |
Ventral tegmental area (circuit 5) | Corticotropin-releasing factor |
Central nucleus of amygdala (circuit 6) | Corticotropin-releasing factor |
BNST (circuit 7) | Norepinephrine |
Nucleus accumbens shell (circuit 8) | Dynorphin |
Habenula (circuit 9) | Acetylcholine |
Central nucleus of amygdala (circuit 10) | Neuropeptide Y |
Central nucleus of amygdala (circuit 11) | Endocannabinoids |
Preoccupation/anticipation | |
Prefrontal cortex (circuit 12) | Glutamate |
Prefrontal cortex (circuit 13) | γ-aminobutyric acid |
Hippocampus (circuit 14) | Glutamate |
Basolateral amygdala (circuit 15) | Glutamate |
BNST (circuit 16) | Corticotropin-releasing factor |
BNST (circuit 17) | Norepinephrine |
Insula (circuit 18) | Corticotropin-releasing factor |
BNST=bed nucleus of the stria terminalis.
Incentive salience
Drugs of abuse have a profound effect on the response to previously neutral stimuli to which the drugs become paired. This phenomenon, called conditioned reinforcement, can be defined as when a previously neutral stimulus reinforces or strengthens behaviours through its association with a primary reinforcer and becomes a reinforcer in its own right. Such cues can be contextual and predictive, and the process of conditioned reinforcement entails not only approach to salient cues but also instrumental responding to turn on the cues, in view of their own rewarding (conditioned reinforcing) properties. As such, conditioned reinforcement as a construct preceded and laid the foundation for incentive salience.55 Incentive salience can be defined as motivation for rewards derived from both one’s physiological state and previously learned associations about a reward cue that is mediated by the mesocorticolimbic dopamine system.56 Both conditioned reinforcement and incentive salience provide constructs for what underlies cue-induced drug seeking, self-administration behaviours, and, conceivably, the transition to habit-like compulsive drug seeking.
The neurobiology that underpins these processes has received much research interest. One particularly influential series of studies in non-human primates indicated that midbrain dopamine cells initially fired in response to a novel reward. After repeated exposure, the neurons stopped firing during predictable reward delivery and instead fired when they were exposed to stimuli that were predictive of the reward.57 A new reward—and subsequently the cues associated with the reward—triggered phasic dopamine cell firing and activation of dopamine D1 receptors, which are necessary for conditioning to occur (table (table2,2, circuits 1–4).58 This process allows previously neutral stimuli to become endowed with incentive salience and strengthens the learned association with repeated exposure to the cues, which creates strong motivation to seek a reward.
Phasic dopamine signalling induced by drug administration can also trigger neuroadaptations in basal ganglia circuits. Here, drug-induced phasic dopamine release triggers the ability of drug-paired cues to increase dopamine levels. Activation of the ventral striatum leads to the recruitment of striatal-globus pallidalthalamocortical loops that engage the dorsal striatum, resulting in habit formation45 and triggering what is hypothesised to underlie compulsive responding for drugs (table (table2,2, circuits 3 and 4).59 Key synaptic changes involve glutamate-modulated N-methyl-D-aspartate (NMDA) receptors and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors in glutamatergic projections from the prefrontal cortex and amygdala to the ventral tegmental area and nucleus accumbens (table (table2,2, circuit 4)32,43,46.
The ability of conditioned cues to recruit these circuits augments progression through the addiction cycle and helps explain intense desire for the drug (ie, craving) and compulsive use when individuals with addiction are exposed to drug cues or stressful environments that are linked with negative emotional states. Conditioned cues within the incentive salience process appear to drive dopamine signalling to maintain motivation to take the drug, even when its pharmacological effects lessen.57,60,61 By contrast, parallel impairments in executive function that are mediated by the prefrontal cortex might be linked to changes in tonic dopamine cell firing that result in lower but more stable dopamine levels in the dopamine projections to the prefrontal cortex.62,63 This mechanism implicates lower-affinity D1 receptors, which stimulate cyclic adenosine monophosphate (cAMP) signalling, as being involved in both acute drug reward and conditioning, because both induce spikes in dopamine release. By contrast, D2 receptors, which inhibit cAMP signalling, are stimulated by both phasic and tonic dopamine and are not deemed necessary for drug reward.40,64 D3 receptors, which are high-affinity dopamine receptors and co-localise in the nucleus accumbens with D1 receptors, are also associated with drug-seeking behaviour.65
Neurobiological mechanisms of the withdrawal/negative affect stage
The withdrawal/negative affect stage consists of key motivational elements, such as chronic irritability, emotional pain, malaise, dysphoria, alexithymia, states of stress, and loss of motivation for natural rewards. Across all major drugs of abuse, this stage is characterised in laboratory animals by elevations in reward thresholds (ie, decreased reward) during withdrawal. In animal models of the transition to addiction, elevations in brain reward thresholds occur that temporally precede and are highly correlated with escalation in drug intake.20 During acute and protracted withdrawal from chronic administration of all drugs of abuse, increases in stress and anxiety-like responses also occur that contribute greatly to the malaise of abstinence and protracted abstinence (table (table11).66 Human brain imaging studies have reported decreases in the sensitivity of brain reward circuits to stimulation by natural rewards during the withdrawal/negative affect stage.
Chronic drug exposure-induced neurochemical changes in systems that are implicated in acute drug reward are called within-system neuroadaptations. Within-system neuroadaptations can be defined as those in which “the primary cellular response element to the drug…adapt[s] to neutralize the drug’s effects; persistence of the opposing effects after the drug disappears… produce[s] the withdrawal response.”68 Such changes include decreases in dopaminergic and serotonergic transmission in the nucleus accumbens during drug withdrawal as measured by in-vivo microdialysis in rats 69 and brain imaging studies in humans, in which amphetamine-induced or methylphenidate-induced striatal dopamine responses are 50% lower in detoxified abusers and 80% lower in active abusers, and accompanied by lower self-reports of the drug’s rewarding effects relative to non-drug-abusing controls.25, 35, 61, 70 Other observed changes include increases in μ opioid receptor responsivity during opioid withdrawal,71, 72 and decreases in GABAergic and increases in NMDA glutamatergic transmission in the nucleus accumbens.73, 74 Differential regional changes in nicotinic receptor function in the nucleus accumbens and ventral tegmental area in nicotine, alcohol, and other addictions have also been reported, implicating α4β2 nicotinic receptor subtypes.19,44 Such decreases in reward system function might persist in the form of long-term biochemical changes that contribute to the clinical syndrome of acute withdrawal and protracted abstinence and could also explain the loss of interest in normal, non-drug rewards (ie, narrowing of the behavioural repertoire toward drugs and drug-related stimuli; tables tables1,1, ,2,2, circuit 5).
The emotional dysregulation that is associated with the withdrawal/negative affect stage also involves a between-system neuroadaptation, in which neuro-chemical systems other than those involved in the positive rewarding effects of drugs of abuse are recruited or dysregulated by chronic activation of the reward system.68 Both the hypothalamic-pituitary-adrenal axis and brain stress system mediated by CRF are dysregulated by the chronic administration of all major drugs with dependence or abuse potential, with a common response of elevated adrenocorticotropic hormone, corticosterone, and amygdala CRF during acute withdrawal.20,75 As tolerance and withdrawal develop, brain stress systems, such as CRF, norepinephrine, and dynorphin, are recruited in the extended amygdala and contribute to the development of negative emotional states in withdrawal and protracted abstinence (tables (tables1,1, ,2,2, circuits 6–8).20,22 As a result, the concept of anti-reward was developed,2 based on the proposal that opponent processes that are a general feature of biological systems also act to limit reward.4 Multiple circuits are likely to contribute to the hypothesised opponent-like processes. During acute withdrawal from all drugs of abuse, CRF increases in the extended amygdala (tables (tables1,1, ,2,2, circuit 6). Importantly, CRF receptor antagonists block both the anxiety-like, stress-like effects of drug withdrawal and excessive drug taking during compulsive drug seeking in animals. Equally compelling, κ opioid receptor antagonists, when injected into the nucleus accumbens shell, can block the development of compulsive drug seeking (tables (tables1,1, ,2,2, circuit 8).76–78 Decreases in the release of dopamine in the nucleus accumbens can also be driven by increases in the activity of the dynorphin-κ opioid receptor system in the ventral striatum and possibly increases in the activity of CRF in the ventral tegmental area, which contributes to the negative emotional state associated with withdrawal and protracted abstinence.20,21
The lateral habenula plays a key role in mediating and encoding aversive states.79 Numerous studies indicate that the habenula is one of the regions that control the decreases in dopamine neuron firing in the ventral tegmental area that are associated with failure to receive an expected reward.80,81 This hypothesis is also consistent with the finding that α5 nicotinic acetylcholine receptors in the habenula appear to modulate aversive responses to large doses of nicotine48 and, along with habenular α2 receptors, nicotine withdrawal (tables (tables1,1, ,2,2, circuit 9).82
Anti-reward circuits are engaged as neuroadaptations during the development of addiction, producing aversive or stress-like states. These aversive states are manifest when the drug is removed during acute withdrawal but also during protracted abstinence.2 Thus, the within-system and between-system construct could be equally valid for the preoccupation/anticipation stage. The combination of decreases in reward function and increases in stress function in the motivational circuits of the ventral striatum, extended amygdala, and habenula is a powerful trigger of negative reinforcement that contributes to compulsive drug-seeking behaviour and addiction. Decreases in reward can be driven by overactivation of the habenula or overactivation of the dynorphin system in the ventral striatum, both of which can decrease dopamine neuron firing. Increases in stress-like states and increased responsivity to stressors could be driven by the recruitment of CRF in the amygdala and other extrahypothalamic stress systems67.
Compulsive-like habits might also derive from the so-called dark side of the withdrawal/negative affect stage. In a study of patients with obsessive-compulsive disorder, patients showed enhanced avoidance habits compared with controls following overtraining on a shock avoidance task, providing support for a habit account of obsessive-compulsive disorder.83 Whether a deficient inhibitory process in prefrontal regions or overactive goal-directed actions and habit processes in the basal ganglia mediate this effect remains to be determined, but the enhancement of avoidance habits provides another basis for the contribution of activation of the stress axis to compulsive-like responding.
Endogenous anti-stress systems also appear to buffer the brain stress systems and influence vulnerability to the development and perpetuation of addiction. Key neurotransmitters that act in opposition to brain stress systems include neuropeptide Y, nociceptin, and endocannabinoids (tables (tables1,1, ,2,2, circuits 10 and 11).20,28,29,49,84 In particular, with chronic drug exposure, neuro-adaptations in the endocannabinoid system, which modulates the response of the brain to stress,85 might contribute to the enhanced stress reactivity in addiction. Indeed, human brain imaging studies have reported reductions of cannabinoid CB1 receptors in cannabis abusers and alcoholics.86,87 Thus, the development of enduring aversive emotional states,88 mediated by overactivation of the stress and anti-reward systems or underactivation of the anti-stress systems, might contribute to the crucial problem in drug addiction of chronic relapse, in which individuals with addiction return to compulsive drug taking long after acute withdrawal.2
Neurobiological mechanisms of the preoccupation/anticipation stage
The preoccupation/anticipation stage has long been hypothesised to be a key element of relapse in humans, and defines addiction as a chronic relapsing disorder. Although this stage has often been linked to the construct of craving, craving in itself has been difficult to measure in human clinical studies and does not always correlate with relapse.89 Nevertheless, the stage in which the individual reinstates drug-seeking behaviour after abstinence remains a focus for identifying neurobiological mechanisms of relapse and the development of medications for treatment.
Executive control over incentive salience is essential to maintain goal-directed behaviour and the flexibility of stimulus–response associations. In rats, the prefrontal cortex (mainly prelimbic cortex, and some infralimbic cortex) sends glutamatergic projections directly to mesocortical dopamine neurons in the ventral tegmental area, thus exerting excitatory control over dopamine cell firing and dopamine release in the prefrontal cortex90 (for correspondence with humans, see figure figure2).2). Given that ventral tegmental area dopamine cells project heavily to the basal ganglia, this frontal cortex glutamatergic projection might contribute to the development of incentive salience. Glutamatergic projections from the prefrontal cortex to the caudate and ventral striatum also modulate the control of the striatal-pallidal-thalamo-cortical system through both direct (D1 receptor-mediated) and indirect (D2 receptor-mediated) pathways. Thus, the prefrontal cortex is in a good position to regulate incentive salience and conditioned behaviour when a salient cue is presented to the individual.92,93
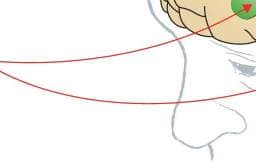
Figure 2: Correspondence between rat and human brain regions relevant to the addiction process
Rats are commonly studied to unveil the neurobiological mechanisms of addiction because they have a well characterised central nervous system whose neurochemical and molecular pathways in subcortical areas correspond reasonably well to those in humans. ACC=anterior cingulate cortex. PL=prelimbic cortex. IL=infralimbic cortex. OFC=orbitofrontal cortex. INS=insula. dlPFC=dorsolateral prefrontal cortex. vlPFC=ventrolateral prefrontal cortex. vmPFC=ventromedial prefrontal cortex. DS=dorsal striatum. GP=globus pallidus. NAc=nucleus accumbens. BNST=bed nucleus of the stria terminalis. CeA=central nucleus of the amygdala. HPC=hippocampus. Modified with permission from George and colleagues (2012),50 Koob and colleagues (2014),91 and the National Academy of Sciences. Evidence from rodent studies suggests that drug-induced reinstatement is mediated by the circuit that links the prelimbic prefrontal cortex to the ventral striatum94 (for correspondence with humans, see figure figure2;2; tables tables1,1, ,2,2, circuits 12 and possibly 13). In rats, neurotransmitter systems that are involved in drug-induced reinstatement involve a glutamatergic projection from the prelimbic prefrontal cortex to the nucleus accumbens that is modulated by dopamine activity through D1 and D2 receptors in the frontal cortex. Cue-induced reinstatement also involves a glutamatergic projection from the prelimbic prefrontal cortex, basolateral amygdala, and ventral subiculum to the nucleus accumbens, and dopamine modulation in the basolateral amygdala and dorsal striatum (tables (tables1,1, ,2,2, circuits 14 and 15).51,52,95 By contrast, the stress-induced reinstatement of drug-related responding in animal models appears to depend on the activation of both CRF and norepinephrine in elements of the extended amygdala (ie, central nucleus of the amygdala and bed nucleus of the stria terminalis; tables tables1,1, ,2,2, circuits 16 and 17)22,33,53,96–98 and the ventral tegmental area (tables (tables1,1, ,2,2, circuit 9). Protracted abstinence, largely described in alcohol dependence models, appears to involve overactive glutamatergic and CRF systems.99,100 Based on animal studies using rodents, increases in activity of the prefrontal glutamatergic system could be hypothesised to elicit a strong glutamatergic response that mediates craving-like responses during the preoccupation/anticipation stage (tables (tables1,1, ,2,2, circuits 10 and 11).
In humans, cue-induced craving appears to involve activation of similar circuits, in which cues that are associated with drugs and elements of non-drug addictions produce activation of the prefrontal cortex, including the dorsolateral prefrontal cortex, anterior cingulate gyrus, and medial orbitofrontal cortex.101–105 Imaging studies have also revealed that cues associated with cocaine craving increase dopamine release in the striatum, amygdala, and prefrontal cortex (anterior cingulate and medial orbitofrontal cortex) and opioid peptides in the anterior cingulate and frontal cortex.9,31,106,107 Indeed, imaging studies have revealed baseline decreases in orbitofrontal (medial and lateral) and anterior cingulate (ventral and dorsal) function and dopamine function during dependence, but reactivation of dopamine and reward system function during acute craving episodes.108
Concomitant with prefrontal activation of a craving system that is mediated by glutamate, human imaging studies have reported deficits in executive function that are reflected by decreases in frontal cortex activity that interfere with decision-making, self-regulation, inhibitory control, and working memory,109 and might involve disrupted GABAergic activity in the prefrontal cortex (table (table2,2, circuit 13).50 For example, alcoholics exhibit impairments in the maintenance of spatial information, disruption of decision-making, and impairments in behavioural inhibition. Indeed, Volkow and colleagues36 and Goldstein and Volkow14 have described a syndrome that is characterised by excessive salience to drug-paired cues, decreases in the responsiveness to non-drug rewards, and decreases in the ability to inhibit maladaptive behaviour. Such frontal cortex-derived executive function disorders in addiction have been linked to deficits in the ability of behavioural treatments to effect recovery.110
Cravings for food, cocaine, and nicotine are also related to middle insular function.111–113 Tobacco smokers with damage to the insula (but not control smokers with extra non-insular lesions) were able to stop smoking easily and without experiencing cravings or relapse.114 The insula appears to have an interoceptive function that integrates autonomic and visceral information with emotion and motivation, thus providing conscious awareness of these urges.54 Brain lesion studies suggest that the ventromedial prefrontal cortex and insula are necessary components of the distributed circuits that support both emotional115 and moral116 decision making. Consistent with this hypothesis, imaging studies have reported differential activation of the insula during craving, possibly reflecting interoceptive cues and hypothesised to involve CRF activation (table (table 2,2, circuit 18).54,117,118 Accordingly, the reactivity of this brain region has been suggested to serve as a biomarker to help predict relapse.119
The capacity to inhibit prepotent responses is a major contributor to an individual’s vulnerability to addiction because it modulates the ability to avoid inappropriate behaviours.120,121 Such a conceptual inhibition system involves a widely distributed and more complex prefrontal cortex–subcortical circuitry. Positron emission tomography studies have uncovered substantial reductions of D2 receptor availability in the striatum in individuals with addiction that persist for months after protracted detoxification, which have also been observed in preclinical studies in rodents and non-human primates with repeated drug exposure.122 These low levels of striatal D2 receptors have been associated with decreases in baseline glucose metabolism (ie, a marker of brain function) in the prefrontal cortex (including dorsolateral prefrontal cortex, anterior cingulate gyrus, and medial orbitofrontal cortex),35,123,124 impulsivity in methamphetamine abusers,125 and compulsive cocaine administration in rodents.15 Consistent with this hypothesis, the converse is also possible. Methamphetamine-dependent individuals with striatal D2 receptor availability within the normal range had better treatment outcomes, potentially reflecting a greater ability to acquire new reward-related behaviours and the pursuit of healthy goals.126
Two opposing systems can therefore be postulated: a Go system and a Stop system. The Go system could drive craving and engage habits via the basal ganglia.105,127 For example, steeper delayed discounting and cocaine craving in cocaine dependence is associated with increased connectivity in the network linking the medial prefrontal cortex and anterior cingulate cortex with the ventral striatum and in the network linking the insula with the dorsal striatum.128 Activation of the dorsal anterior cingulate is also associated with fear conditioning in post-traumatic stress disorder.129 The Stop system could control assessment of the incentive value of choices and suppression of affective responses to negative emotional signals.130–132 Under this framework, a Stop system would inhibit the Go craving system and stress system. For example, in post-traumatic stress disorder there is substantial evidence of hypoactivity in the ventromedial prefrontal cortex and hyperactivity in the central nucleus of the amygdala that reflects an inhibitory connection between the ventromedial prefrontal cortex and central nucleus of the amygdala.133,134 Indeed, successful cognitive inhibition of craving in cocaine abusers has been associated with inhibition in the right medial orbitofrontal cortex and activation in the right inferior frontal cortex.135
Molecular and genetic treatment targets within brain circuits associated with addiction
The neuroplastic changes outlined previously are triggered and sustained by molecular and cellular adaptations that can presumably also interact with genetic and environmental vulnerability to addiction. In the binge/intoxication stage, both signal transduction mechanisms and changes in gene transcription have been identified. For example, chronic exposure to a wide variety of abused drugs upregulates cAMP formation, cAMP-dependent protein kinase A (PKA) activity, and PKA-dependent protein phosphorylation in the nucleus accumbens. Numerous interventions that tonically activate nucleus accumbens cAMP/PKA signalling promote escalations in drug self-administration or compulsive-like drug-seeking behaviour, and the upregulation of a postsynaptic Gs/cAMP/PKA signalling pathway in the nucleus accumbens might constitute a critical neuroadaptation that is central to the establishment and maintenance of the addicted state.136,137
These changes in signal transduction can trigger longer-term molecular neuroadaptations via transcription factors that modify gene expression. A well-characterised example is that chronic exposure to various drugs of abuse increases the activation of cAMP response element binding protein in the nucleus accumbens and deactivates it in the central nucleus of the amygdala. Introduction of cAMP response element binding protein in the nucleus accumbens decreases the reinforcing value of natural and drug rewards, and this change plausibly contributes to withdrawal/negative affect stage-related decreases in reward pathway function, which leave a drug-abstinent individual in an amotivational, dysphoric, or depressed-like state.138,139 These substance-related changes in susceptibility to negative emotional states might begin early: alcohol use during adolescence might lead to epigenetic modifications that alter amygdalar gene expression and dendritic density, increasing susceptibility to anxiety-like behaviours and alcohol ingestion in adulthood.140
Substance-induced changes in transcription factors can also produce competing effects on reward function.141 For example, repeated substance use activates accumulating levels of ΔFosB, and animals with elevated ΔFosB exhibit exaggerated sensitivity to the rewarding effects of drugs of abuse, leading to the hypothesis that ΔFosB might be a sustained molecular trigger or switch that helps initiate and maintain a state of addiction.141,142 Next-generation sequencing methods have shown that repeated cocaine exposure leads to histone modifications that act in a combinational fashion to create chromatin signatures that strikingly alter pre-mRNA splicing, an effect that is necessary for the expression of cocaine-induced conditioned place preference.143
The heightened risk of cue-induced cocaine seeking (incubation of craving) that occurs during prolonged withdrawal (protracted abstinence in humans) has provided new insights into the molecular basis of vulnerability to relapse. During incubation in animal models, AMPA-type glutamate receptors are recruited. Specifically, the infralimbic medial prefrontal cortex-to-nucleus accumbens shell circuit recruits calcium-permeable AMPA receptors,144 and the prelimbic medial prefrontal cortex-to-nucleus accumbens core circuit recruits non-calcium-permeable AMPA receptors. The blockade of metabotropic glutamate 1 receptors in the nucleus accumbens blocks cue-induced incubation reinstatement and cocaine-primed reinstatement; as a result, these receptors are potential therapeutic targets.145,146
The kinase mammalian (mechanistic) target of rapamycin complex 1 (mTORC1) belongs to the phosphoinositide 3-kinase (PI3K)-related kinase (PIKK) family and plays a key role in the dendritic translation of synaptic proteins. As such, mTORC1 plays a major role in the molecular mechanisms associated with learning and memory147 and is involved in several brain disorders, including epilepsy, Parkinson’s disease, Alzheimer’s disease, and addiction.148,149 Exposure to drug and alcohol cues activates the mTORC1 pathway in the hippocampus, frontal cortex, nucleus accumbens, and amygdala.149 Even more compelling, blockade of the mTORC1 pathway systemically blocked the reconsolidation of cocaine memories,150 and blockade of mTORC1 in the amygdala blocked the reconsolidation of alcohol memories;151 these results suggest a potential molecular target for the treatment of relapse.
Heritability of addictions is 40–60%, much of which is caused by genetic variations that affect underlying neurobiological mechanisms, and thus is consistent with there being common pathways to different addictions.152 Genes have been identified that convey vulnerability at all three stages of the addiction cycle, and salient candidates are discussed within this conceptual framework. However, a more comprehensive analysis is beyond the scope of this Review. In the binge/intoxication stage, several genes have been identified in animals as key to drug responses, and their modifications strongly affect drug self-administration.37,153–155 Notably, in animal models, dopamine D1 receptor knockout rats will not self-administer cocaine37 and μ opioid receptor knockout mice will not show the rewarding effects of opioids.153 In humans, only a few specific genes have been identified with polymorphisms (alleles) that either predispose an individual to or protect an individual from drug addiction,156 but the number is growing. For example, genome-wide association studies have implicated two acetylcholine receptors, the α4 nicotinic receptor subunit157 and α5 nicotinic receptor subunit,158 in the vulnerability to nicotine dependence and a single-nucleotide polymorphism associated with regulating the trafficking and gating properties of AMPA-selective glutamate receptors (CNIH3) in opioid dependence.159
Some of the polymorphisms associated with vulnerability for the binge/intoxication stage interfere with drug metabolism. For example, specific alleles for genes that encode alcohol dehydrogenase (ADH1B) and acetaldehyde dehydrogenase (ALDH2; enzymes involved in the metabolism of alcohol) are reportedly protective against alcoholism.160 Intriguingly, an evolutionarily conserved GABA synthesis pathway mediated by aldehyde dehydrogenase 1a1 (ALDH1a1) has been identified in the ventral tegmental area.161 GABA co-release is modulated by binge-like doses of alcohol administered to mice, and diminished ALDH1a1 leads to enhanced alcohol consumption and preference. Similarly, polymorphisms in the genes for cytochrome P450 2A6 and 2D6 (enzymes involved in nicotine and opioid metabolism, respectively) are reportedly protective against nicotine addiction.162
From the perspective of the withdrawal/negative affect stage, in humans, two single-nucleotide poly morphisms in the CRF1 receptor gene (CRHR1) were associated with binge drinking in adolescents and excessive drinking in adults.163 Moreover, homozygosity at one of these single-nucleotide polymorphisms (rs1876831, C allele) was associated with heavy drinking in relation to stressful life events in adolescents.164
From the perspective of the preoccupation/anticipation stage, a genetic knock-in mouse that biologically recapitulates a common human mutation in the gene for fatty acid amide hydrolase (C385A [Pro129Thr], rs324420) presented a reduction of fatty acid amide hydrolase expression associated with the variant allele that selectively enhanced fronto-amygdala connectivity and fear extinction learning and decreased anxiety-like behaviours; this result suggests another possible molecular genetic target for neuroplasticity in the preoccupation/anticipation and withdrawal/negative affect stages.165
Although such approaches do not guarantee that these genes convey vulnerability in the human population, they provide viable candidates for exploring the genetic basis of endophenotypes that are associated with addiction.
Developmental exposure as a key component of vulnerability for drug and alcohol use disorders
Normal developmental processes might result in higher risk for drug use at some stages of the lifecycle than others. Experimentation, as well as the process of addiction, most often starts in adolescence,166 a period during which the brain undergoes important developmental changes.167 Beginning in preadolescence and continuing into the mid-20s, cortical grey matter volumes reduce, which reflects a normal pruning process; white matter volume increases over the course of adolescence, reflecting increases in connectivity, including axonal extension and myelination.168 Drug exposure during adolescence is associated with more chronic and intensive use and greater risk of a substance use disorder than is initiation at older ages.167,169–172 Normal adolescent-specific behaviours (e.g., risk-taking, novelty-seeking, and high sensitivity to peer pressure) increase the propensity to experiment with legal and illegal drugs,167 which might reflect the incomplete development of brain regions (eg, myelination of frontal lobe regions) that are involved in the processes of executive control and motivation.168 Heavy alcohol use during adolescence is associated with a range of neurobehavioural sequelae, including impairments in visuospatial processing, attention, and memory,173,174 and adolescents who had engaged in episodes of heavy drinking presented faster declining volumes in some neocortical grey matter regions and smaller increases in regional white matter volumes than did non-drinking adolescents.175 Additionally, voluntary binge drinking in animal models during adolescence reduced the density of myelinated axons in the medial prefrontal cortex.176
Environmental factors that have been consistently associated with the propensity to self-administer drugs include structural factors (eg, low socioeconomic status and associated lack of social support systems), proximal factors (eg, parental drug use and dependence, as per the DSM-IV, poor quality of parenting, parental depression, and sibling and peer influences), and more distal factors (eg, drug availability, school, neighbourhood characteristics, advertising, and the media).177,178 Stress is also a common feature that increases the risk for drug abuse. The mechanisms that are responsible for stress-induced increases in vulnerability to drug use and relapse in those who are addicted are not yet well understood, but evidence suggests that the stress-responsive neuropeptide CRF is involved through its effects in the amygdala and hypothalamic-pituitary-adrenal axis.20
Relevance to behavioural addictive disorders
The three stages of the addiction cycle are pervasive and form common domains in non-drug addictions, also known as process addictions,179 such as pathological gambling, binge-eating disorder, compulsive buying, and internet addiction disorder.180 Non-drug addictions elaborate self-regulation failures similarly to drug addictions, with transitions from impulsivity to compulsivity and a chronic relapsing trajectory. Similar brain mechanisms, particularly with regard to reward deficits, stress sensitisation, and impaired inhibitory control, have been observed in behavioural or process addictions, as well as decreases in striatal dopamine D2 receptors associated with reduced activity in prefrontal cortices,181 deficits in the Go craving system and Stop inhibition systems,182,183 and dysregulation of the stress axis.184
Implications for medication development
Our contention is that the field of the neurobiology of addiction has excellent and validated animal models and well-developed clinical models that, combined with the neurocircuitry analysis herein, will provide a unique approach to medication development that emphasises excessive incentive salience, the loss of brain reward function, and the gain of stress function that drive negative reinforcement (ie, the dark side of addiction) and the dysregulation of executive function, all of which are key components that drive compulsive drug seeking.13,185 Advances in diagnosis that reflect such an endophenotype or research domain criterion approach will synergistically combine with advances in neurobiology to promote novel pharmacotherapeutic, brain stimulation (ie, transcranial magnetic stimulation and direct electrical stimulation), and behavioural treatments.186
Conclusions and future directions
We elaborate here a heuristic framework based on the behavioural and imaging phenotypes of addiction as three stages linked by three functional domains that are mediated by three major neurobiological circuits (basal ganglia, extended amygdala, and prefrontal cortex) and numerous microcircuits of neuroplasticity. We outline 18 neurochemically defined mini circuits that can independently or interactively load the outputs of the major common neurobiological circuits to produce incentive salience and compulsive-like habits, negative emotional states of low reward and excessive stress, and compromised executive function. Identification of these molecular and neurochemical loads on the circuitry provides key information about vulnerability, resilience, treatment, and recovery from addiction, as well as information on how different drugs of abuse enter the overall addiction cycle. Non-drug addictions, such as pathological gambling, have a similar behavioural phenotype that fits into the three-stage cycle. Preliminary imaging data also suggest common neurobiology. Key questions that remain are what genetic factors load these mini circuits, how the environment conveys epigenetic influences on these circuits, and how these circuits recover or do not recover with abstinence and treatment. Resolving these questions should provide biomarkers for prevention, behavioural windows of development for prevention, novel behavioural and pharmaceutical treatments, and ultimately a neurobiological understanding of recovery.