Abstract
Tobacco smoking is highly addictive and causes respiratory disease, cardiovascular disease and multiple types of cancer. Electronic-cigarettes (e-cigarettes) are non-combustible tobacco alternatives that aerosolize nicotine and flavouring agents in a propylene glycol-vegetable glycerine vehicle. They were originally envisaged as a tobacco cessation aid, but whether or not they help people to quit tobacco use is controversial. In this review, we have compared and contrasted what is known regarding the effects of nicotine on the lungs vs. the effects of nicotine in the brain in the context of addiction. Critically, both combustible tobacco products and e-cigarettes contain nicotine, a highly addictive, plant-derived alkaloid that binds to nicotinic acetylcholine receptors (nAChRs). Nicotine's reinforcing properties are primarily mediated by activation of the brain's mesolimbic reward circuitry and release of the neurotransmitter dopamine that contribute to the development of addiction. Moreover, nicotine addiction drives repeated intake that results in chronic pulmonary exposure to either tobacco smoke or e-cigarettes despite negative respiratory symptoms. Beyond the brain, nAChRs are also highly expressed in peripheral neurons, epithelia and immune cells, where their activation may cause harmful effects. Thus, nicotine, a key ingredient of both conventional and electronic cigarettes, produces neurological effects that drive addiction and may damage the lungs in the process, producing a complex, multilevel pathological state. We conclude that vaping needs to be studied by multi-disciplinary teams that include pulmonary and neurophysiologists as well as behaviourists and addiction specialists to fully understand their impact on human physiology.
Introduction
E-cigarettes are non-combustible electronic nicotine delivery devices that contain nicotine in a liquid vehicle of propylene glycol and vegetable glycerine along with a broad range of flavours (Stratton et al. 2018). The e-cigarette liquid (e-liquid) is then heated in a battery-powered device and inhaled. People use tobacco and e-cigarettes to get nicotine to the brain: conventional tobacco products and newer e-cigarette devices are highly efficient nicotine delivery systems. Tobacco smoking is declining because of multiple cultural factors, including bans on tobacco advertising and flavoured products and increased taxes (Wakefield et al. 2011; McNeill et al. 2017). In contrast, vaping is less regulated and has been increasing in popularity in many countries since these restrictions do not yet apply. Thus, a current challenge is to keep up with cultural changes both from scientific and public health perspectives in order to provide evidence-based research for the legislature. Indeed, research in this area is struggling to keep pace with a dynamic marketplace, escalating use and changing cultural perceptions. Whether or not e-cigarettes are safer than conventional tobacco smoking has been hotly contested, and their effects on neuronal function and underlying behaviours associated with e-cigarette use remain unclear. Similarly, their effects on the pulmonary system, the body's first point of exposure with inhaled e-liquids, is also poorly understood, although evidence is emerging that e-cigarette use is not as safe as previously thought (Eltorai et al. 2019; Gotts et al. 2019) and is associated with increased rates of pulmonary disease (Bhatta & Glantz, 2020). In this review, we shall discuss the effects of e-cigarette-delivered nicotine on the brain and lungs, in an attempt to better understand how nicotine can contribute to the altered physiology seen with e-cigarette exposure.
E-cigarettes as an alternative to smoking tobacco
The effects of e-cigarettes are not well understood and represent a poorly characterized health risk (Dinakar & O'Connor, 2016; Gotts et al. 2019). E-cigarettes are widely perceived as a safer alternative to tobacco smoking (Gravely et al. 2014; McMillen et al. 2015; Filippidis et al. 2017). However, public health specialists have offered differing opinions regarding their safety and there are conflicting data regarding their usefulness as smoking cessation tools (Hartmann-Boyce et al. 2016; Jankowski et al. 2017). Public Health England recently doubled down on previous advice that e-cigarettes are 95% safer than smoking and that people should switch from tobacco to e-cigarettes (East et al. 2018). This advice was based on a panel review that estimated that the risk of vaping is <5% that of smoking based on the number of known cancer-causing agents (Nutt et al. 2014). This advice has recently been questioned (Eissenberg et al. 2020) and a recent European Respiratory Society task force noted that the long-term health effects of vaping are unknown and there is no evidence that e-cigarettes are safer than tobacco (Bals et al. 2019). In multiple countries, the popularity of e-cigarettes amongst youths has led to increased nicotine use, whilst tobacco smoking rates amongst similar age groups is flat or declining (Cullen et al. 2018; Hammond et al. 2019), and there is also concern that e-cigarette-mediated nicotine adoption by youths may lead to long-term nicotine dependency.
Nicotine overview
Nicotine is an alkaloid that is secreted from plants of the nightshade family as an insecticide. Nicotine binds to nicotinic acetylcholine receptors (nAChRs), ubiquitously expressed ligand-gated cation channels that are related to GABA and 5-HT receptors (Albuquerque et al. 2009; Benowitz, 2009). They are composed of five subunits and each subunit has four transmembrane domains and an extracellular N-terminal ligand binding site (Fasoli & Gotti, 2015). They are classified as either α or β based on the presence or absence of an extracellular cysteine domain, respectively, and there are eight human α-subunits (α2–7, 9, 10) and three β-subunits (β2–4) (Dani, 2015). Combinations of these subunits produce numerous nAChRs with variable ligand binding affinities and physiological roles (Dani, 2015). Acetylcholine is the physiological ligand for these receptors, and binding of acetylcholine or exogenous nicotine opens the channel to allow influx of cations (Na+, K+ and Ca2+). The channels subsequently close and become desensitized (Dani, 2015). The reinforcing effects of nicotine are primarily mediated by the α7 and α4β2 nicotinic receptors in the mesolimbic reward pathway, whilst α7 receptors are commonly expressed in the lung and in immune cells (Gahring & Rogers, 2006; Zdanowski et al. 2015). nAChR activation causes excitation of neighbouring neurons, resulting in rapid synaptic transmission (Fasoli & Gotti, 2015). However, these neuronal excitatory effects are downregulated by chronic exposure to low concentrations of nicotine (Dani, 2015). nAChRs (α4β2) rapidly desensitize upon nicotine binding, and upregulation/resensitization leads to nicotine craving (Rose, 2007; Benowitz, 2010; England et al. 2015). Whilst nicotine has been studied on its own, its contribution towards tobacco-induced lung pathology is less well understood, since it tends to be studied along with the other chemicals in cigarette smoke rather than in isolation. It is tacitly assumed that free base nicotine will behave similarly when added directly, in an e-cigarette aerosol or in tobacco smoke. However, there is a knowledge gap in the field and this relationship has not been extensively tested. Moreover, Juul-type e-liquids contain nicotine salt, rather than nicotine free base, and its effects on the airways, brain and other organs, as well as its efficacy relative to freebase nicotine, are poorly understood.
Connecting nicotine, the lung and the brain
Addiction to nicotine drives the repeated exposure to electronic nicotine vapor in humans (Fig. 1). Indeed, exposure to nicotine is reinforcing and can lead to repeated cycles of intake culminating in the need for regular consumption and withdrawal symptoms during periods of abstinence (Markou, 2008). One of the primary goals of addiction research is to understand how nicotine or other drugs of abuse augment or impair cellular functions to produce long-lasting maladaptive changes to brain circuitry that promote addiction. However, nicotine passes through the lungs and cardiovascular system before it reaches the brain. Thus, it is important to understand the detrimental effects of nicotine from a systems perspective. The rest of the body, and the lungs in particular, is chronically exposed to nicotine, which leads to activation of peripheral nAChR. Thus, future studies will need to examine the interplay between peripheral and central effects of nicotine: for example, (1) how nicotine can directly alter cell signalling pathways both centrally and peripherally to change gene/protein expression, and (2) how peripheral alterations can trigger maladaptive changes in specific regions of the lungs and brain.
Figure 1. Nicotine intake alters lung homeostasis and acts in the brain to promote addiction
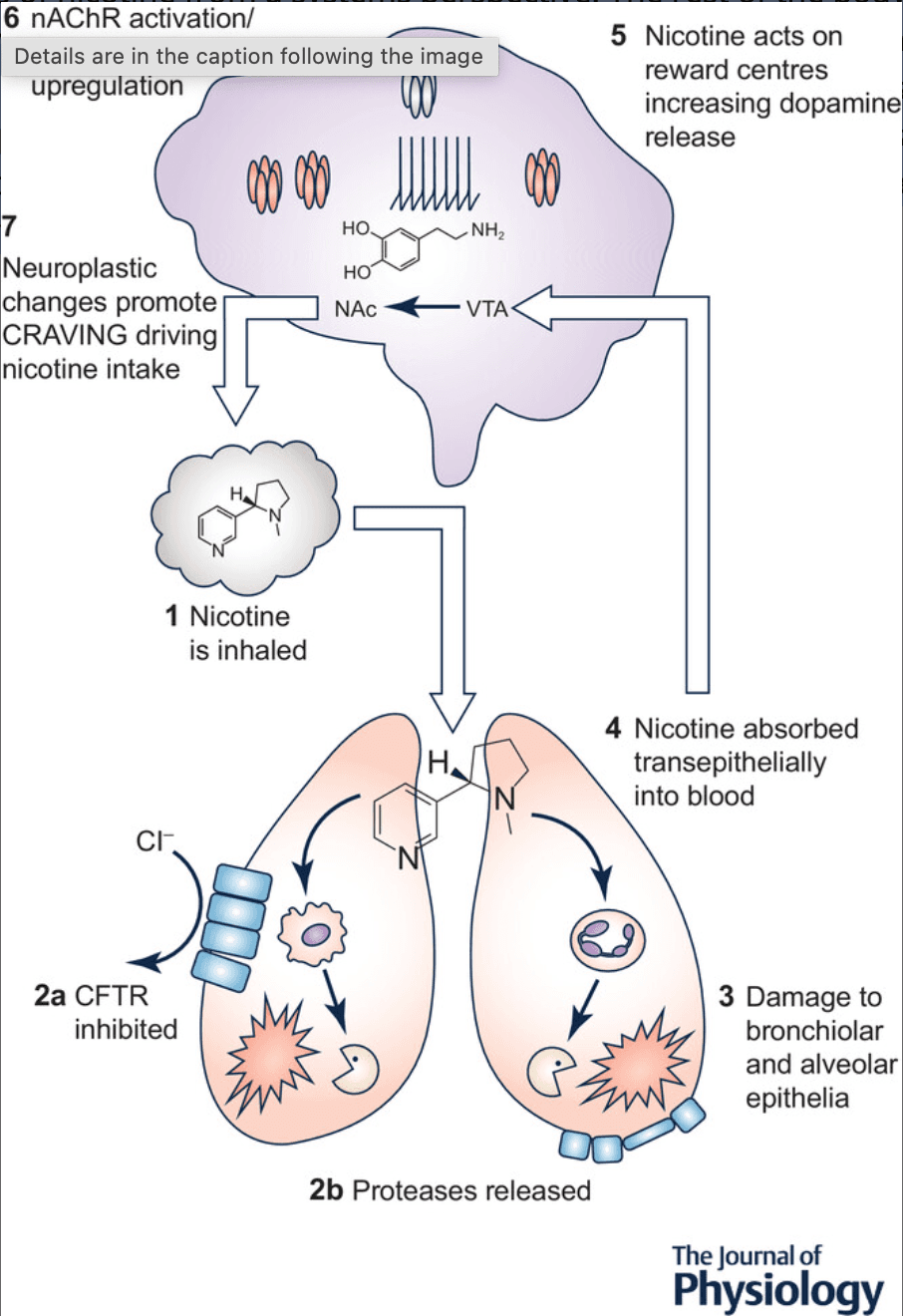
(1) Nicotine is inhaled into the lungs where it equals or exceeds 50 μm in the airway surface liquid. (2) Nicotine can activate nAChRs in the lung. (2a) Nicotine may inhibit the CFTR Cl− channel, potentially leading to dehydration. (2b) Nicotine stimulates protease release from immune cells, which may lead to (3) lung damage (e.g. bronchiectasis and emphysema). (4) Nicotine is absorbed systemically where it then crosses the blood–brain barrier. (5) Nicotine acts on the reward centres in the brain (ventral tegmental area, VTA; nucleus accumbens, NAc) to release dopamine. (6) nAChR densensitization and subsequent upregulation promotes (7) neuroplastic changes associated with craving leading to repeated nicotine uptake and more exposure into the lungs (1).
The effects of nicotine on neuronal adaptations and behaviour
The behaviours associated with nicotine exposure and dependence are mediated by cellular adaptations as the brain responds to repeated cycles of exposure and withdrawal. With prolonged use, these central adaptations can lead to near-permanent changes in neurons and neuronal networks (Markou, 2008). A number of brain regions have been identified as targets of nicotine-induced plasticity, including the amygdala and the mesolimbic reward pathway (Adinoff, 2004). The mesolimbic reward pathway originates in the ventral tegmental area (VTA) with dopamine projections into the nucleus accumbens. Nicotine, as well as drugs like cocaine, amphetamine and alcohol, increases mesolimbic dopamine signalling by enhancing the excitability of VTA dopamine cells. Nicotine acts at nAChRs expressed in VTA dopamine neurons as well as on local GABAergic interneurons and afferent terminals (Pidoplichko et al. 2004) to alter dopamine neuron excitability through direct actions on dopamine cell bodies as well as alterations in local GABAergic and glutamatergic transmission (Mansvelder & McGehee, 2000, 2002; Mansvelder et al. 2002). Nicotine-induced augmentation of mesolimbic dopamine signalling is particularly sensitive to mechanisms regulating the excitability of VTA dopamine cell bodies (Pidoplichko et al. 2004). Substantial evidence indicates that the motivational effects of nicotine are largely mediated through the VTA. For example, lesions of VTA dopamine neurons or intra-VTA infusion of nAChR antagonists decreases nicotine self-administration (Corrigall et al. 1992, 1994), and mice and rats will self-administer nicotine directly into the VTA in a manner sensitive to nicotinic acetylcholine and dopamine receptor blockade (Ikemoto et al. 2006). These studies indicate that motivational and rewarding effects of nicotine are mediated by increased excitation of VTA dopamine cells (Mansvelder & McGehee, 2000, 2002; Mansvelder et al. 2002). In contrast to acute exposure, chronic nicotine is associated with a diminished dopaminergic state. VTA dopamine cell firing (Liu & Jin, 2004), tonic and phasic dopamine release and extracellular dopamine levels in striatal regions of rodents and non-human primates (Takahashi et al. 1998; Rahman et al. 2004; Domino & Tsukada; Zhang et al. 2012), and dopamine metabolite levels in cerebrospinal fluid from human smokers (Geracioti et al. 1999) are all decreased following chronic nicotine exposure. These deficits in dopamine transmission are thought to contribute to decreased brain reward function (Epping-Jordan et al. 1998) and nicotine withdrawal symptoms including depressed mood, decreased arousal and sleep disturbances (Takahashi et al. 1998; Rahman et al. 2004; Domino & Tsukada, 2009; Zhang et al. 2012). Chronic exposure-related deficits in dopamine function partially result from nicotine-induced disruptions in excitatory and inhibitory mechanisms regulating midbrain dopamine cell activity (Takahashi et al. 1998; Rahman et al. 2004; Domino & Tsukada, 2009; Zhang et al. 2012). In addition to effects on the central nervous system, nicotine has significant effects on the parasympathetic nervous system, including increased locomotion and decreased body temperature (Javadi-Paydar et al. 2019). Thus, irrespective of its source, inhaled nicotine produces significant cellular neuronal adaptations.
Nicotine pharmacodynamics
After e-cigarette inhalation, blood nicotine levels typically peak at ∼120 nm (St Helen et al. 2016). In contrast, little has been done to study nicotine levels in the lung. Unlike for blood and urine, measuring nicotine levels in the lung is a technically challenging endeavour that requires sampling of the airway surface liquid (ASL) that lines the lung lumen either by performing bronchoscopy and obtaining bronchoalveolar lavage or by obtaining sputum. Further, the nature of these techniques makes repeat measurements difficult, so it is harder to develop comprehensive lung nicotine pharmacodynamic profiles. This impediment notwithstanding, we have previously measured nicotine levels in sputum of smokers and vapers (Clunes et al. 2008; Ghosh et al. 2019). Here, we corrected for any dilution factors by measuring levels of lung urea, since this biomarker is typically at equilibrium between blood and sputum. Using this approach, we found that sputum nicotine levels were ∼30 μm for smokers (Clunes et al. 2008). We found that sputum nicotine levels in vapers were ∼50 μm (Ghosh et al. 2019). Moreover, since these measurements were made ∼30 min after vaping, we may be markedly underestimating the amount of nicotine seen by the lung, since nicotine is likely transepithelially absorbed in an exponential fashion. E-liquids contain between 3 and 18 mg ml−1 of nicotine, which equates to between ∼18 and 112 mm. Thus, if we are seeing ∼50 μm nicotine after ∼30 min, the initial nicotine concentration seen by the lung is likely much higher and possibly in the millimolar range. Clearly, more studies are needed to fully determine the impact of e-cigarette device type and e-liquid type (including nicotine concentration), as well as variations in subject topography on lung nicotine levels.
The effects of nicotine on the lung
Airway epithelia
Airway epithelia line the lung lumen (Whitsett & Alenghat, 2015). They help modulate ASL volume/composition, secrete mucins, secrete cytokines that can trigger leukocyte infiltration and also form a barrier against invading pathogens (Shaykhiev & Crystal, 2013). Airway epithelia are deranged following chronic tobacco smoke exposure and undergo significant changes in gene and protein expression that lead to a loss of barrier function, goblet cell metaplasia and altered inflammatory status (Ghosh et al. 2015; Strzelak et al. 2018). We have found that vaping causes significant changes to the airway epithelial proteome that are distinct from the changes seen in smokers. These changes were accompanied by an altered physical appearance of the airways, and vapers’ airways had a distinct reddish colour that was indicative of erythema and increased friability (Ghosh et al. 2018). However, further studies will be required to determine the impact of nicotine on these changes. The effects of vaping on the lungs of never-smokers have been studied. Never smokers inhaled a nicotine-containing e-liquid and saw significant changes in gene expression (Staudt et al. 2018). In a second study, in which subjects only inhaled propylene glycol/vegetable glycerine, the changes appeared to be much smaller (Song et al. 2020), suggesting that changes may have been nicotine-dependent. Thus, inhaling nicotine via e-cigarettes may contribute to the changes seen in vapers’ airways.
ASL provides an appropriate environment for immune cell function and is a source of proteases and protease inhibitors (Hiemstra, 2015; Taggart et al. 2017). Maintenance of ASL hydration is critical for efficiently clearing mucus out of the lungs. Indeed, in both cystic fibrosis and chronic obstructive pulmonary disease (COPD), dysfunctional cystic fibrosis transmembrane conductance regulator (CFTR)-mediated anion secretion contributes to ASL dehydration that leads to mucus plugging, chronic infection and inflammation, and lung damage (i.e. bronchiectasis) (Collawn & Matalon, 2014; Ghosh et al. 2015). In COPD airways, mucus dehydration inversely correlates with a decline in lung function and also is a predictor of mortality (Hogg et al. 2004; Anderson et al. 2015). More recently, nicotine has been shown to inhibit CFTR function leading to decreased Cl− secretion, decreased ciliary beating and decreased airway hydration (Fig. 1; Garcia-Arcos et al. 2016; Chung et al. 2019; Lin et al. 2019). Whilst the mechanism of nicotine-dependent CFTR inhibition is not fully understood, we have previously found that CFTR is inhibited by elevations in cytoplasmic Ca2+ (Rasmussen et al. 2014; Patel et al. 2019). This causes CFTR dephosphorylation (Marklew et al. 2019), and we speculate that nicotine-dependent Ca2+ influx through nAChRs likely inhibits CFTR through a similar process, indicating a mechanistic link between nicotine intake and epithelial ion channel dysfunction.
Pulmonary immune cells
Alveolar macrophages are resident innate immune cells that phagocytose, and secrete cytokines, chemokines and growth factors (Gordon & Read, 2002; Rubins, 2003; Phipps et al. 2010; Lawal, 2018). nAChRs are highly expressed in pulmonary immune cells (Gahring & Rogers, 2006). For example, α7 nAChR knockout mice show a blunted pulmonary response to cigarette smoke exposure (Gahring et al. 2017). Alveolar macrophages exhibit functional heterogeneity/plasticity (Gordon & Taylor, 2005; Hao et al. 2012). Notably, long-term activation of macrophages without resolution of inflammation can cause airway damage. Conversely, downregulation of macrophage function can lead to immunosuppression, airway infection and inflammation-associated damage (Simonin-Le Jeune et al. 2013). In the healthy lung, alveolar macrophages make up >90% of the resident immune cells. However, some neutrophils (∼3–5% of the total cell count) are also present, and this number can change drastically during infection and/or inflammation. For example, in COPD lungs, neutrophils become the predominant cell type in the lung (Jasper et al. 2019). Scott et al. (2018) found that e-liquid condensate exposure increased apoptosis and necrosis in alveolar macrophages, in a nicotine-dependent fashion. Similarly, phagocytosis was impaired by nicotine-containing e-liquids, suggesting that innate defence in alveolar macrophages may be impaired. Neutrophils can also phagocytose and release cytokines/chemokines. However, neutrophils are also prone to lysis and can release their intracellular contents, including proteases and DNA into the lung, which can contribute to lung damage and increased mucus/sputum viscosity, respectively (Kaplan & Radic, 2012). Neutrophil lysis and the formation of DNA-containing neutrophil extracellular traps (NETs) is triggered by elevations in extracellular Ca2+. Nicotine activation of nAChRs, via its ability to elevate cytoplasmic Ca2+, can also induce NETosis/neutrophil lysis (Lee et al. 2017).
Proteases including neutrophil elastase, which as its name suggests, is derived from neutrophils, and macrophage-derived matrix metalloproteases (MMP-2 and MMP-9) are normally expressed in the lung, where they are involved in tissue repair and regeneration (Greene & McElvaney, 2009). However, when chronically upregulated, these proteases cause lung damage (emphysema and bronchiectasis; Fig. 1; Nadel, 2000; Skrzydlewska et al. 2005; Abboud & Vimalanathan, 2008; Fischer et al. 2011). They can also degrade antimicrobial proteins (Webster et al. 2018) and cleave epidermal growth factor receptor leading to altered cellular communication and increased mucin expression (Greene & McElvaney, 2009). We have previously conducted research bronchoscopies on healthy vapers and smokers. We found that neutrophil elastase and macrophage-derived matrix metalloproteases (MMP-2 and MMP-9) were significantly elevated in vapers’ lungs, to the same extent as seen in smokers (Ghosh et al. 2019). Studies of freshly isolated alveolar macrophages and peripheral blood neutrophils from healthy non-smokers revealed that nicotine caused a dose-dependent increase in cytosolic Ca2+ and protease release from both cell types (Ghosh et al. 2019). Macrophages were found to be more sensitive to nicotine than neutrophils and their EC50 for protease release was ∼40 nm, which was below the amount of nicotine (∼50 μm) measured in vapers’ sputum. In keeping with these findings, lung protease levels were also elevated in vapers who were never-smokers. In mice, vaping with nicotine and propylene glycol–vegetable glycerine also caused emphysema (Garcia-Arcos et al. 2016). Given the firmly established link between proteases and lung damage, the potential risk to vapers’ lungs from nicotine-induced proteolysis cannot be overstated.
Future studies and conclusions
To improve understanding of the impact of e-cigarette/nicotine exposure on the lung and brain, greater integration between clinical researchers and behaviourists studying human e-cigarette-mediated nicotine intake and laboratory researchers studying animal and cellular models of e-cigarette/nicotine exposure is required. One future challenge will be to accurately reproduce human nicotine intake and exposure levels in the laboratory. Moreover, exposure levels will likely differ depending on the organ system being studied, with higher levels of exposure seen in the lungs than in the brain or other systems. Differentiating the peripheral and central effects of nicotine, and how these distinct but parallel processes interact, potentially through inflammatory signalling or neuroimmune activation, is also an important area of future study.
Animal models will be essential in studying the effects of vaping. To date, mice have been exposed to e-cigarette vapour, leading to emphysema, increases in mucin levels and lipid accumulation, amongst other findings (Garcia-Arcos et al. 2016; Ghosh et al. 2018; Madison et al. 2019). However, other animals including large animals whose airways more accurately reflect human airway physiology will be important. For example, sheep have also been exposed to e-cigarette vapour, which causes mucus stasis (Chung et al. 2019). Importantly, animal models will allow for the study of multi-organ pathology. For example, vaping impairs embryo implantation in pregnant mice and alters the development of the offspring (Wetendorf et al. 2019). Vaping also induced fibrosis and caused impaired renal function in mice (Crotty Alexander et al. 2018). Regardless of the animal type or organ studied, challenges facing the field include (1) possible strain-dependent effects, (2) lack of appropriate e-cigarette aerosol exposure regimens, and (3) given the large number of e-liquids that are commercially available (over 7000 and counting) and the dynamic nature of the market place, finding an appropriate e-liquid to use. For example, whilst Garcia-Arcos et al. (2016) found emphysema after vaping mice, Madison et al. (2019) did not, and instead found altered lipid accumulation, which may have been due to strain dependencies or different vape exposure protocols. Similarly, Lee et al. (2018) exposed mice to e-cigarette aerosol for a year, which is a massive time commitment, and during this time, vendors may change the composition of, or discontinue e-liquids. This field is still in its infancy, and standard test e-liquids and standard exposure protocols are urgently needed, much akin to the Kentucky research cigarettes and standard exposure protocols used with conventional cigarettes.
In conclusion, electronic vaporization of nicotine likely promotes the same addictive behaviours as nicotine exposure through conventional means, resulting in increased chronic/repeated use, which will have deleterious effects in the brain and lung (Fig. 1). The lung has evolved to be highly resilient and it typically takes decades of tobacco smoke exposure before pathology emerges (i.e. COPD or lung cancer). Thus, even though e-cigarettes have already been shown to induce changes to multiple regions of the lung (Gotts et al. 2019), what we are seeing is likely to be only the tip of the iceberg. Indeed, whilst incidences of lung cancer and COPD spiked to match peak tobacco smoking usage in the last century and are now falling, there was a lag time between the two events, with tobacco use declining before disease. Decades from now, we may see a new e-cigarette-dependent spike in lung disease, so it is critical that addiction researchers and those studying the lung and other systems exposed to nicotine work together on this problem in order to avoid a late 21st century nicotine/e-cigarette addiction and lung disease epidemic.