Abstract
Mental health symptoms secondary to trauma exposure and substance use disorders (SUDs) co-occur frequently in both clinical and community samples. The possibility of a shared aetiology remains an important question in translational neuroscience. Advancements in genetics, basic science, and neuroimaging have led to an improved understanding of the neural basis of these disorders, their frequent comorbidity and high rates of relapse remain a clinical challenge. This project aimed to conduct a review of the field’s current understanding regarding the neural circuitry underlying posttraumatic stress disorder and SUD. A comprehensive review was conducted of available published literature regarding the shared neurobiology of these disorders, and is summarized in detail, including evidence from both animal and clinical studies. Upon summarizing the relevant literature, this review puts forth a hypothesis related to their shared neurobiology within the context of fear processing and reward cues. It provides an overview of brain reward circuitry and its relation to the neurobiology, symptomology, and phenomenology of trauma and substance use. This review provides clinical insights and implications of the proposed theory, including the potential development of novel pharmacological and therapeutic treatments to address this shared neurobiology. Limitations and extensions of this theory are discussed to provide future directions and insights for this shared phenomena.
INTRODUCTION
Research focused on trauma and posttraumatic stress disorder (PTSD) dates back to 1889, when Pierre Janet, a prominent French psychiatrist, published L’Automatisme Psychologique, an early attempt to describe how the mind processes traumatic events[1]. Janet argued that patients suffering from dissociation and hysteria had unresolved traumatic memories and that this subconscious experience was routed in the physical effects of past negative experiences. When an individual experiences a traumatic event, they are overwhelmed with intensely negative emotions and are unable to accurately process and remember details surrounding the event[2]. The traumatic experience dissociates from conscious awareness. Janet believed that the individual would relive the memory of the trauma in fragmented pieces, such as emotional states, somatic conditions, visual images, or behavioral re-enactments. Janet was the first to identify dissociation as the crucial psychological technique involved in a variety of post-traumatic symptoms[1].
Decades of research on understanding the psychological and biological effects of trauma have provided support for many of Janet’s early observation[1]. Psychological distress following a traumatic event can manifest through a variety of symptoms including anxiety and exaggerated fear as well as anhedonia, dysphoria, anger, aggression, or dissociation[3]. The 5th edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) provides a description of seven different diagnoses which all relate to exposure to a prominent or stressful event, the most prevalent of these diagnoses are Acute Stress Disorder and PTSD. Criteria for PTSD include direct exposure to or indirect witnessing of a traumatic or stressful event, as well as the presence of intrusive symptoms, negative mood, dissociation, avoidance and/or arousal. These symptoms must cause clinically significant distress or impairment and must not be attributable to the effects of a substance, a medical condition, or a brief psychotic disorder[3]. Patients with PTSD must also display at least one or more symptom across each category (intrusive, avoidance, negative alterations in cognition and mood, alterations in arousal and reactivity). Interpersonal trauma is one of the most common criteria A events and is more likely to lead to poorer functional outcomes[3].
Given the diversity and complexity of individual responses to trauma, it has been difficult to fully understand the neurobiological substrates involved in PTSD. The majority of basic neuroscience and neuroimaging research on PTSD has focused primarily on two areas; the effect of stress on sympathetic nervous system functioning and the impact of trauma on frontal striatal brain circuitry[4]. Although such research has begun to shed light on the adverse neurobiological effects of repeated stress, many patients continue to suffer from the effects of trauma and are resistant to empirically validated treatments for PTSD[5]. An emerging area of research that may help to further elucidate the neurobiological mechanism of trauma relates to the important role of reward circuitry in translational models of PTSD. After providing an overview of animal and clinical research on the biological effects of PTSD, this article will review recent research that suggests an important role for the brain’s reward pathway in understanding the neurobiological effects of complex trauma.
THE PHYSIOLOGY AND NEUROBIOLOGY OF PTSD
The human stress response and PTSD
During stress, the sympathetic nervous system prepares an individual for action while the pituitary-adrenocortical system dampens initial physiological aspects of arousal[6]. The hypothalamic (HPA) axis releases corticotrophin releasing factor (CRF) which stimulates the release of cortisol from the adrenal cortex, and increases the release of catecholamine neurotransmitters within several regions of the brain[7]. Catecholamines have an integral role in the adaptive response to stress through the breakdown of glycogen, the suppression of the insulin release, and increased functioning of the cardiovascular system[6]. Increased levels of norepinephrine (NE) and epinephrine (EPI) during stress result in increased neuronal activity in limbic areas such as the amygdala, and hypothalamus, and decreased activation of cortical areas involved in higher-order cognitive functioning[8].
For individuals at risk for developing PTSD, traumatic experiences can alter the normal functioning of the sympathetic nervous system. Many of the core symptoms of PTSD reflect a state of hyperarousal including exaggerated startle response, initiating and maintaining sleep and poor concentration[9]. Patients with PTSD demonstrate exaggerated sympathetic nervous system responses including tachycardia and skin conductance during acute stress and increased sensitivity of the HPA axis[10]. Compared to healthy controls, patients with PTSD have reduced baseline cortisol levels, and increased levels of CRF[11]. Several studies have demonstrated that patients with PTSD have higher urinary secretion levels of NE, and EPI, compared to controls and that neurotransmitters levels correlate with the severity of self-reported PTSD symptoms[10,12]. Previous PTSD studies have reported related abnormalities in sensory processing including deficits in the P50 and P300 evoked potential component[13].
Abnormalities in HPA axis and neurotransmitter function can alter neural circuitry both structural and functional changes of, especially in brain circuits integral to affective and cognitive processing. While high levels of cortisol enhance the formation of emotional memories (mediated by the increased amygdala function) and facilitates fear conditioning, high levels of cortisol upon trauma exposure decreases hippocampus function, resulting in memory and learning deficits[14]. Structural changes occur as well; high levels of stress can result in dendritic hypertrophy of the prefrontal cortex (PFC), and dendritic remodeling of the amygdala[15].
Between stimulus and response
How does the brain process sensory information that may be perceived as a threat? LeDoux was among the first to demonstrate the underlying neural circuitry of fear (Figure 1) For example, imagine that a door slams shut in the middle of the night, waking you up from sleep. The initial sensory information about the threat is relayed to the thalamus and then quickly sent to the amygdala, activating the stress response and generates an immediate reaction. The body begins to sweat, a diaphoretic response that precipitates a rapid response. The hippocampus and PFC process contextual information about the stimulus by providing reasoning (perhaps it was a windy day) and episodic memory (a slamming door has never caused you any harm) that dampens the stress response and allow an individual to relax, returning to baseline. The signal from the thalamus to the amygdala is rapid, while signals from the hippocampus and PFC are transmitted with less velocity[16]. Recent translational research has demonstrated how structural, chemical and functional differences in each of these brain areas evolve in the neurobiology of PTSD.
Given the role of intrusive memories in PTSD, many have speculated that hippocampal dysfunction is a critical component of the underlying neurobiology. The hippocampus has a high concentration of corticosteroid receptors that are involved in the termination of the stress response through the negative feedback of the HPA axis[17]. Both animal and human studies have demonstrated that high levels of stress can damage the hippocampus, resulting in memory impairments[18]. During an acute stress response, high levels of cortisol can diminish dendritic branching in the hippocampus while augmenting neurogenesis in the amygdala, enhancing the emotional salience of the event, but impairing memory functioning[15,19]. When compared with healthy controls, patients with PTSD demonstrate impairments in short-term memory and some studies (but not all) have demonstrated reduced hippocampal volumes in patients with PTSD[20]. Patients with PTSD often have difficulty describing details related to traumatic events and some studies have correlated hippocampal regional cerebral blood flow with PTSD symptom severity[21,22].
The amygdala receives input from the thalamus as well as sensory processing regions of the neocortex, and transmits signals to autonomic brain structures, thereby playing a critical role in both the sympathetic and parasympathetic stress response[23,24]. In human studies, the amygdala has been demonstrated to have a critical role in processing emotional stimuli, and in the formation of emotionally salient memories[25]. It also has a role in fear conditioning, whereby a neutral conditioned stimulus is associated with an unconditioned stressful stimulus. Exposure to the conditioned stimulus initiates the stress response, activates the amygdala, and engages the autonomic nervous system[26]. Lesion of the amygdala interrupts fear learning and the conditioned response in animals[27,28]. Human neuroimaging studies have confirmed the involvement of this region in fear learning, conditioning and extinction[29,30]. Compared to healthy controls, patients with PTSD demonstrate increased activation of the amygdala when presented with trauma-related cues, as well as when presented with unrelated affective stimuli[31,32]. Amygdala response in patients with PTSD has been found to correlate with self-reported symptom severity[33,34].
Alterations in prefrontal cortical (PFC) activity may help to link the role of memory impairment in PTSD as well as increased amygdala activation during the stress response[20]. In patients with PTSD, repeated exposure to trauma damages these neural structures. The ability to extinguish emotional memories involves the ventromedial PFC as well as the amygdala while extinguishing conditioned fear involves the anterior cingulate cortex and the amygdala[24,35]. Activation of the medial PFC also occurs when inhibiting fearful responses or altering one’s perception of a negative emotional event and therefore decreased functioning of the PFC may explain why patients with PTSD exhibit difficulties in extinguishing fearful memories[36,37].
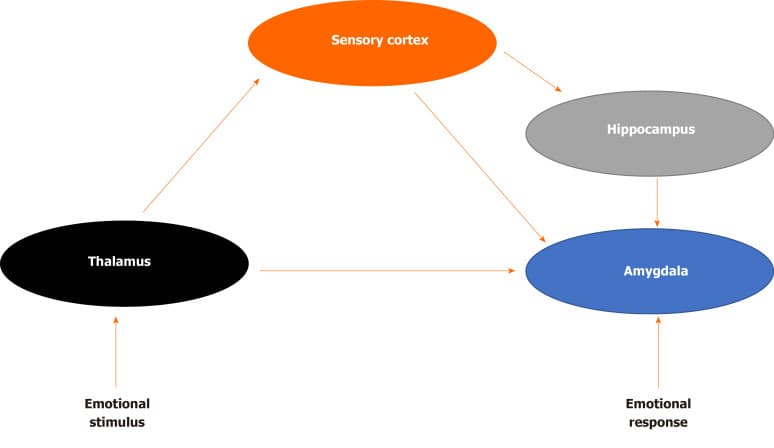
Figure 1. Fear brain circuitry.
COMORBIDITY OF PTSD AND SUBSTANCE USE DISORDERS
Clinical observations, case reports and epidemiological studies first suggested high rates of comorbidity between PTSD and substance use disorders (SUD), prompting translational research examining the possibility of overlapping neural mechanisms. Many studies have demonstrated a high comorbidity of PTSD with drug addiction in both clinical and community samples[38,39]. Approximately 36% to 50% of those that meet criteria for SUD also meet criteria for lifetime PTSD, and those with PTSD predictably have a history of drug abuse or dependence[40]. Comorbidity of these disorders is associated with negative treatment outcomes, increased risk for chronic diseases, and poorer functionality[41]. Co-twin studies have also demonstrated a link between childhood trauma and the later development of SUD[42].
Robinson and Berridge[43] proposed a model of addiction that demonstrates how repeat drug use disrupts normal reward processing. The Incentive-Sensitization Theory postulates that although increased pleasure is initially an important part of addiction behavior, regular substance use increases an individual’s sensitivity to drug cues, causing them to become hyper-responsive to drug cravings, even in the absence of pleasure[43]. This hyper-sensitization produces goal-directed behavior (“wanting”) not only in the absence of subjective pleasure, but also in the absence of consciously being aware of “wanting”. Recent research has supported this theory, demonstrated that substance use can alter brain reward circuitry[43].
BRAIN REWARD CIRCUITRY AND SUD
Data from several studies suggest that the reward circuit of the brain (the mesocortical dopamine pathway) provides a common molecular pathway with which to understand SUD. The mesolimbic pathway involves connections between the ventral tegmental area (VTA), the nucleus accumbens (NAc) in the ventral striatum, and the PFC. The mesolimbic dopamine reward circuit controls the reinforcing and rewarding effects related to food, sex, and social interaction[44]. Drug-induced adaptations in mesolimbic dopamine system (includes common adaptations to many different drugs) mediate changes in reward mechanisms that in part underlie addiction — including tolerance, dependence-withdrawal, sensitization, and relapse. Drug-induced adaptations include regulation of dopamine and opioid systems (mechanisms of tolerance and sensitization), regulation of glutamate systems (influences drug-related memories), upregulation of the cyclic adenosine monophosphate (cAMP) pathway, and transcription factor cAMP Response Element-Binding (CREB) protein, (mechanisms of drug tolerance, dependence, and withdrawal), structural changes in VTA neurons (influences drug tolerance), and structural changes in NAc neurons (influences drug sensitization)[45].
Many studies show that dopamine and accumbens neurons often become most active in anticipation of rewards, not during the reward phase, and also activated by the anticipation of aversive stimuli and events[46,47]. The role of the mesolimbic DA system is to increase the salience of stimuli and events associated with activation of the system. Stimuli are imbued with salience, making them “wanted” incentive stimuli. Alcaro et al[46] have theorized the role of the mesolimbic pathway as driving an organism toward “seeking” behaviors, searching to boost the salience of activities that are life-promoting while avoiding those that are harmful to survival. This proposed role is not only congruent with evidence of the importance of SUD, but also a mechanism that explains the connection of the VTA to the hippocampus, amygdala, and PFC, all of which have been implicated in the neurobiology of PTSD.
OVERLAPPING NEUROBIOLOGY OF PTSD AND SUD
Both animal models and clinical studies of PTSD have noted deficits in reward processing consistent with hypofunctionality of the mesolimbic pathway. Upon exposure to chronic stress, animal models demonstrate reduced striatal dopaminergic activity and decreased reward-seeking behavior that mimic symptoms of anhedonia experienced by PTSD patients[48]. Corral-Frias et al[49] utilized a novel animal model of PTSD to demonstrated that inactivation of the VTA can lead to long-term behavioral changes that mimic the clinical symptoms of PTSD. Inactivation of the VTA through either a dopamine antagonist or bilateral dissection can also lead to chronic changes in baseline VTA dopaminergic cell firing, demonstrating that trauma can lead to long-term alterations of the reward pathway. Evidence of deficits in the brain reward and reinforcement circuits in patients with PTSD also supports the involvement of the mesolimbic dopamine reward circuit[50,51]. In clinical studies, PTSD patients spend less time engaging in reward-seeking behavior compared to controls, report lower levels of reward expectation and are less satisfied with monetized rewards[52,53]. When compared to healthy controls, patients with PTSD demonstrate reduced bilateral striatal activation when responding to positive to reward gains and reported significantly higher levels of motivational and social deficits[50]. Collectively, these findings suggest a strong overlap in the brain regions involved in both fear processing and addiction. In particular, the VTA, through its connections to the amygdala, hippocampus and PFC, may serve as the common substrate of this overlapping circuitry, explaining the high co-morbidity in PTSD and SUD.
Several rodent models provide converging evidence for the overlapping circuitry of these two disorders, including the dorsal and ventral subdivisions of medial PFC and their respectively outputs to the amygdala and NAc (Figure 2). The prelimbic (PL) cortex projects to the basal (BA) nucleus of the amygdala, which excites the central (CE) nucleus of the amygdala, thereby promoting the expression of conditioned fear[54]. The BA also receives excitatory input from lateral amygdala, which also drives the expression of conditioned fear. The infralimbic (IL) cortex, in contrast, excites a class of GABAergic inhibitory neurons (the intercalated cell masses) which inhibit the CE, thereby promoting extinction of the conditioned fear[55]. PL and IL control drug seeking via their differential projections to the core and shell subdivisions of the NAc. The PL projects to the core, which promotes the expression of drug-seeking behavior. The IL projects to the shell, which also promotes the expression of extinction[56].
Functional magnetic resonance imaging studies provide consistent support for similar networks in human studies of fear and addiction (Figure 3). The dorsal portion of the anterior cingulate cortex is associated with fear expression during conditioning behavioral tasks, and overlaps with proximal regions that are activated when SUD patients respond regarding their craving levels after exposure to cocaine-related cues[57-59]. These findings are congruent with results from positron emission tomography mapping of cerebral blood flow using 15O-labeled water[60]. The ventral medial PFC (vmPFC) is activated during fear extinction recall and during recall of addiction cues in individuals with SUD disorders[57,61]. During states of cocaine craving, the vmPFC is deactivated, suggesting a failure to engage extinction[54]. Collectively, these studies suggest that the vmPFC is homologous to rodent IL, whereas the dorsal regions of anterior cingulate cortex are homologous to rodent PL.
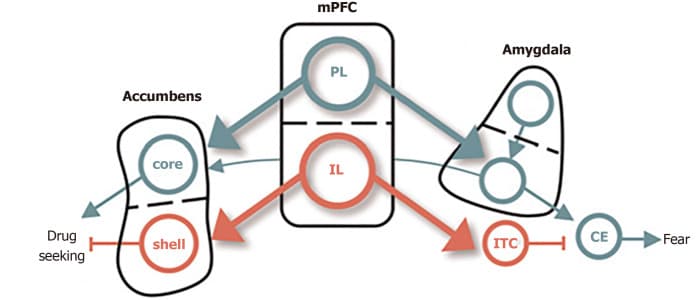
Figure 2. Conditioned fear in cocaine use. The dorsal and ventral subdivisions of medial prefrontal cortex are shown at the center, with their respective outputs to the amygdala controlling fear shown at right, and those to the nucleus accumbens, controlling cocaine, seeking shown at left. Green depicts pathways that activate fear and cocaine seeking. Red depicts pathways that inhibit fear and cocaine seeking[65]. Citation: Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem 2009; 16(5): 279-288. Copyright ©Cold Spring Harbor Laboratory Press 2009. Published by Cold Spring Harbor Laboratory Press[65].
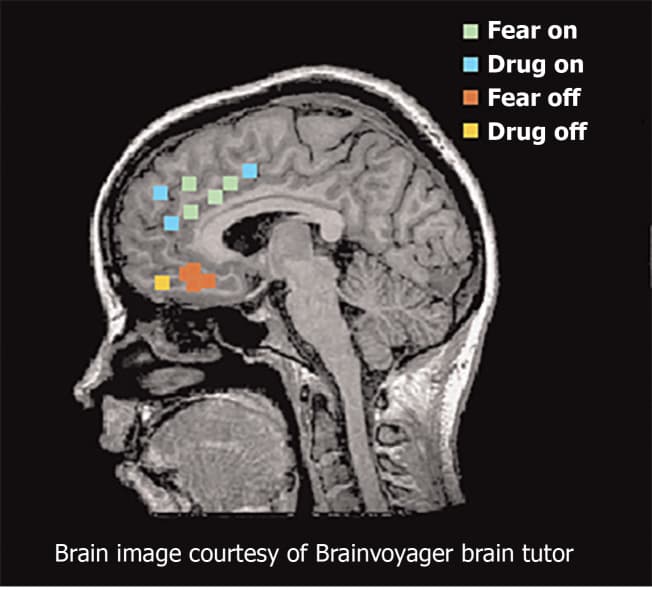
Figure 3. Functional magnetic resonance imaging and positron emission tomography studies of fear and addiction. Green dots represented human dorsal anterior cingulate cortex that correlated with fear expression functional magnetic resonance imaging[65]. Blue dots represent regions that correspond with drug cravings after exposure to cocaine-related cues. Red dots represent regions associated with fear extinction recall. Yellow dots represent regions activated during addiction-related cues. Citation: Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem 2009; 16(5): 279-288. Copyright ©Brain Innovation 2009. Published by Cold Spring Harbor Laboratory Press[65].
CONCLUSION
This review examined evidence in support of a shared neurological origin between PTSD and SUD, in an effort to explain the high rates of comorbidity. It is clear that abnormalities in the PFC and VTA are central to the pathology of both disorders. The VTA is negatively affected during trauma and stress, and results in a decrease of dopaminergic activity and a subsequent alteration in the reward pathway. The PFC is involved in drug seeking behavior as well as the extinction of fear conditioning, playing a role in both addiction and PTSD. This review did not examine the genetic vulnerabilities nor neurodevelopmental pathways that may confer increased risk for either or both disorders, and it remains an important question whether the shared biology reviewed here is due to more distal risk factors, or are a result of one disorder conferring increased risk for the other.
It is important to gain a better understanding of the connection between PTSD and SUD in order to develop improved treatments that target both disorders. Despite shared neurobiology, there are few treatment options that target both, although notably some do exist (e.g., Seeking Safety). Yet many patients are not able to benefit from combined treatment interventions during the earlier stages of substance use recovery, and clinicians often struggle to determine the priority of treatment[62]. Many individuals diagnosed with comorbid PTSD-SUD believe that the outcomes of their disorders are interconnected, yet are not offered treatment for PTSD alongside SUD interventions[63].
Understanding comorbidity may also further prevention efforts, consistent with the “self-medication” hypothesis, as individuals with untreated trauma utilize substance as unhealthy coping mechanisms[64]. Earlier identification, access to care, and treatment of trauma across the lifespan is critical for intervening before the development of SUD or other maladaptive behaviours. Further research must leverage mechanisms between these two disorders, to ensure a more effective and efficient treatment option.