Abstract
Attention-deficit/hyperactivity disorder (ADHD) is a severely impairing neurodevelopmental disorder with a prevalence of 5% in children and adolescents and of 2.5% in adults. Comorbid conditions in ADHD play a key role in symptom progression, disorder course and outcome. ADHD is associated with a significantly increased risk for substance use, abuse and dependence. ADHD and cannabis use are partly determined by genetic factors; the heritability of ADHD is estimated at 70–80% and of cannabis use initiation at 40–48%. In this study, we used summary statistics from the largest available meta-analyses of genome-wide association studies (GWAS) of ADHD (n=53 293) and lifetime cannabis use (n=32 330) to gain insights into the genetic overlap and causal relationship of these two traits. We estimated their genetic correlation to be r2=0.29 (P=1.63×10−5) and identified four new genome-wide significant loci in a cross-trait analysis: two in a single variant association analysis (rs145108385, P=3.30×10−8 and rs4259397, P=4.52×10−8) and two in a gene-based association analysis (WDPCP, P=9.67×10−7 and ZNF251, P=1.62×10−6). Using a two-sample Mendelian randomization approach we found support that ADHD is causal for lifetime cannabis use, with an odds ratio of 7.9 for cannabis use in individuals with ADHD in comparison to individuals without ADHD (95% CI (3.72, 15.51), P=5.88×10−5). These results substantiate the temporal relationship between ADHD and future cannabis use and reinforce the need to consider substance misuse in the context of ADHD in clinical interventions.
Introduction
Attention-deficit/hyperactivity disorder (ADHD) is a neurodevelopmental disorder with a prevalence of 5% in children and adolescents and of 2.5% in adults. It is a severely impairing disorder which impacts significantly on the academic, social, emotional and psychological functioning of an individual and causes high costs for the healthcare system and society.
In addition to the core symptoms of inattention, hyperactivity and impulsivity, comorbid conditions in ADHD cause considerable functional and psychosocial impairments. They also worsen symptom progression, disorder course and outcome. The pattern of psychiatric comorbidity in ADHD is highly heterogeneous and changes substantially across the lifespan. Externalizing disorders are frequently associated with ADHD, with co-occurring substance use disorder (SUD) being common. In fact SUD is more prominent in adulthood and has a prevalence rate of 45% in adult ADHD subjects. Longitudinal and cross-sectional studies show that a diagnosis of ADHD significantly increases the risk for substance use, abuse and dependence in adolescents and adults independently of other psychiatric comorbidity.
Cannabis is the illicit drug most commonly used among individuals with ADHD. Its consumption may lead to the use of other drugs, which in turn can lead to higher rates of ADHD symptoms. The association between ADHD and cannabis use has been reported in cross-sectional and retrospective studies in ADHD patients and in the general population. Prospective studies showed that childhood ADHD is associated with cannabis use and cannabis disorder in adulthood. Particularly, impulsivity and opposition problems during childhood predicted an increased risk of cannabis consumption un adulthood. In addition, individuals with persistent ADHD have shown higher rates of cannabis dependence compared to those with remitted ADHD.
Both ADHD and cannabis use have a highly complex aetiology, implying a combination of genetic and environmental risk factors. The heritability of ADHD is around 70–80% in children and adults; cannabis use initiation has heritability of 48% for males and 40% for females. The aetiology of ADHD and cannabis use can be hypothesized to overlap, and both traits might share an underlying genetic background. However, despite consistent evidence showing that individuals with ADHD may be more prone to consume cannabis, to date no common genetic risk factors or causal links between these traits have been described.
Inferring causality in observational studies is problematic due to confounding, reverse causation and other unknown biases. However, using genetic data, Mendelian randomization approaches may overcome some of these issues and allow causal inference from observational data. Mendelian randomization uses genetic variants robustly associated with an exposure to test whether this exposure causes an outcome, by considering them unconfounded proxies for the exposure. The rationale behind this method is that alleles are passed from parents to offspring randomly, avoiding therefore confounding or reverse causation issues, similar to the allocation of treatments in a randomized controlled trial.
To clarify the nature of the relationship between ADHD and lifetime cannabis use we analysed data from the largest available meta-analyses of genome-wide association studies for these traits and we i) estimated their genetic correlation, ii) undertook a cross-trait analysis to identify shared genetic factors and iii) tested the causal role of ADHD on subsequent cannabis use performing a two-sample Mendelian randomization approach.
Materials and methods
Samples
Summary statistics for ADHD were obtained from the European ancestry subgroup of the Psychiatric Genomics Consortium and iPSYCH (PGC+iPSYCH).This recent meta-analysis of ADHD GWAS, comprises 19 099 cases and 34 194 controls. Summary statistics for the lifetime cannabis use meta-analysis of GWAS, comprising 14 374 cases and 17 956 controls, were obtained from the International Cannabis Consortium (ICC).
Because of sample overlap in two studies between the PGC and ICC samples and to avoid biases, we removed these studies (Spain, 572 cases and 425 controls, and Yale-Penn, 182 cases and 1315 controls) from the PGC+iPSYCH sample in all analyses except for the LD score regression, since this method is not affected by sample overlap. This provided a restricted PGC+iPSYCH sample of 18 345 cases and 32 454 controls.
Quality control and filters applied
As described in, PGC+iPSYCH studies of ADHD imputed their data using the 1000 Genomes Project Phase 3 reference panel, and filtered variants with info score ≤ 0.8, minor allele frequency (MAF) ≤ 1% or N effective ≤ 70%. Each study included in the meta-analysis of lifetime cannabis use from the ICC imputed their data using the 1000 Genomes Project Phase 1 reference panel, excluded indels, removed SNPs with MAF < √5/𝑁, imputation quality scores below 0.6, SNPs present in only one sample and SNPs with alleles or allele frequencies inconsistent with the 1000 Genomes Phase 1 European reference panel (absolute MAF difference > 0.15).
For our analyses, variants with different alleles in PGC+iPSYCH and ICC, with AT/GC alleles or in the HLA region (chromosome 6 and 26 000 000<position<3 3000 000) were removed, and the final number of markers considered was 5 009 020.
SNP-based heritability and genetic correlation between ADHD and lifetime cannabis use
We used single-trait LD score regression to estimate SNP-based heritability for each trait and cross-trait LD Score regression to estimate the genetic correlation between ADHD and lifetime cannabis use considering N effective sample sizes (𝑁𝑒𝑓𝑓𝑒𝑐𝑡𝑖𝑣𝑒=4𝑁𝑐𝑎𝑠𝑒𝑠𝑁𝑐𝑜𝑛𝑡𝑟𝑜𝑙𝑠𝑁𝑐𝑎𝑠𝑒𝑠+𝑁𝑐𝑜𝑛𝑡𝑟𝑜𝑙𝑠). Data for 1 064 988 markers, overlapping the HapMap 3 reference panel used by the LD score regression software were included in this analysis.
Cross-trait analysis
In order to avoid sample overlap, the Spanish and Yale-Penn studies were excluded from the PGC+iPSYCH meta-analysis of ADHD using a weighted difference for the beta and standard error estimates between the meta-analysis and these two studies.
A fixed effect inverse variance weighted meta-analysis across the restricted PGC+iPSYCH results of ADHD and the ICC lifetime cannabis use results was run as the main analysis, and a random effects meta-analysis was run as a sensitivity analysis; both using plink v1.9. software. Clumping of the cross-trait analysis results was performed using the following parameters: r2=0.2, kb=250, p2=0.5, p1=5×10−8. Conditional analyses for top signals in regions previously reported by PGC+iPSYCH for ADHD were undertaken using the GCTA software and an in-house cohort of European ancestry individuals (n=3 719) as reference for LD calculations. Individuals in this cohort were genotyped using the Infinium PsychArray-24 BeadChip, HumanOmni1-Quad BeadChip or HumanOmni2.5 BeadChip platforms (Illumina Inc., San Diego, California, USA) and imputed to the 1000 Genomes Project Phase 1 reference panel. The gene-based analysis was performed using MAGMA. SNPs were assigned to genes if they were within a 10kb window upstream or downstream of the gene. Default MAGMA gene coordinates defined according to NCBI37.3 were used. Mean SNP associations were calculated per gene and gene P-values were obtained using a known approximation of the sampling distribution. LD information was extracted from the 1000 Genomes Project Phase 1 reference panel. This analysis was performed with the cross-trait results, both for the fixed and random effect analyses, as well as with the PGC+iPSYCH data on ADHD and the lifetime cannabis use data from the ICC for comparison. The genome-wide significance threshold for the gene-based analysis was set at P=2.79×10−6 after a Bonferroni correction considering a total of 17 927 genes. Plots were generated using the R package “qqman” and locuszoom.
Sign test
The sign test was undertaken selecting variants associated with ADHD and assessing whether their direction of effect was consistent for cannabis use. Then, variants associated with cannabis use were selected and we assessed whether their direction of effect was consistent in ADHD. The test used was a one sample test of the proportion with Yates’ continuity correction against a null hypothesis of P=0.50 with the “stats” package in R-3.3.3. Strict clumping (r2=0.05, kb=500 and p2=0.5) was applied at different P-value thresholds of 5×10−8, 5×10−7, 5×10−6 and 5×10−5.
Mendelian randomization
The analysis was undertaken in both directions, (i) using ADHD as exposure and lifetime cannabis use as outcome, and (ii) using lifetime cannabis use as exposure and ADHD as outcome. Strict clumping was undertaken in the exposure population with parameters r2=0.05, kb=500 and p2=0.5 using plink v1.9. The following thresholds were used: P<5×10−8 (including 12 variants) and P<5×10−6 (including 72 variants) when using ADHD as exposure, and P<5×10−6 (including 9 variants) and P<5×10−5(including 70 variants) when using cannabis use as exposure, given that no SNPs had P<5×10−8 and only one had P<5×10−7 in the ICC dataset.
For a Mendelian randomization analysis to be valid the following assumptions need to be met: (i) the genetic variant(s) need to be robustly associated with the exposure, (ii) the only way the genetic variant(s) may be associated with the outcome is through the exposure, and (iii) the genetic variant(s) must be independent from unobserved confounders that may influence the exposure and the outcome. We used the inverse-variance weighted (IVW) method as the main analysis to obtain the average effect across genetic variants. This method provides an efficient estimate when all genetic variants are valid instruments (all assumptions are met for all variants). We also ran MR-Egger regression, MR-PRESSO and the weighted median method as sensitivity analyses. MR-Egger regression allows all variants to have pleiotropic effects (when a variant affects the exposure and the outcome independently), violating assumption (ii), as long as an additional, weaker assumption holds: direct pleiotropic effects of the genetic variants on the outcome are distributed independently of the genetic associations with the exposure (Instrument Strength Independent of Direct Effect, InSIDE, assumption). MR-Egger regression measures the average pleiotropic effect across the genetic variants by estimating the intercept and tests whether its value (log OR) is different from zero. MR-PRESSO assumes that at least 50% of the variants are valid instruments, there is balanced pleiotropy and the InSIDE assumption holds; it undertakes a test to detect pleiotropy (global test) and in case of pleiotropy it corrects it by outlier detection and removal. The weighted median method provides a consistent estimate when up to 50% of the genetic variants are invalid instruments (violating assumptions (ii) and/or (iii)). Additionally, we ran heterogeneity tests and repeated analyses removing one genetic variant at a time (leave-one-out analyses). We used “MendelianRandomization” and “TwoSampleMR” packages with R-3.3.3.
The Mendelian randomization causal estimate of the effect of ADHD on cannabis use represents the odds of cannabis use per unit increase in the log OR of ADHD risk. In order to convert the estimate to the odds of cannabis use for ADHD versus non-ADHD we used a method previously described assuming a prevalence of ADHD of 5%.
Results
SNP-based heritability and genetic correlation between ADHD and lifetime cannabis use
The SNP-based heritability estimated was 26% for ADHD and 9% for lifetime cannabis use (Sup Table 1). We found strong evidence of SNP-based genetic correlation between the two conditions (rg=0.29, se=0.068, P=1.63×10−5).
Cross-trait analysis
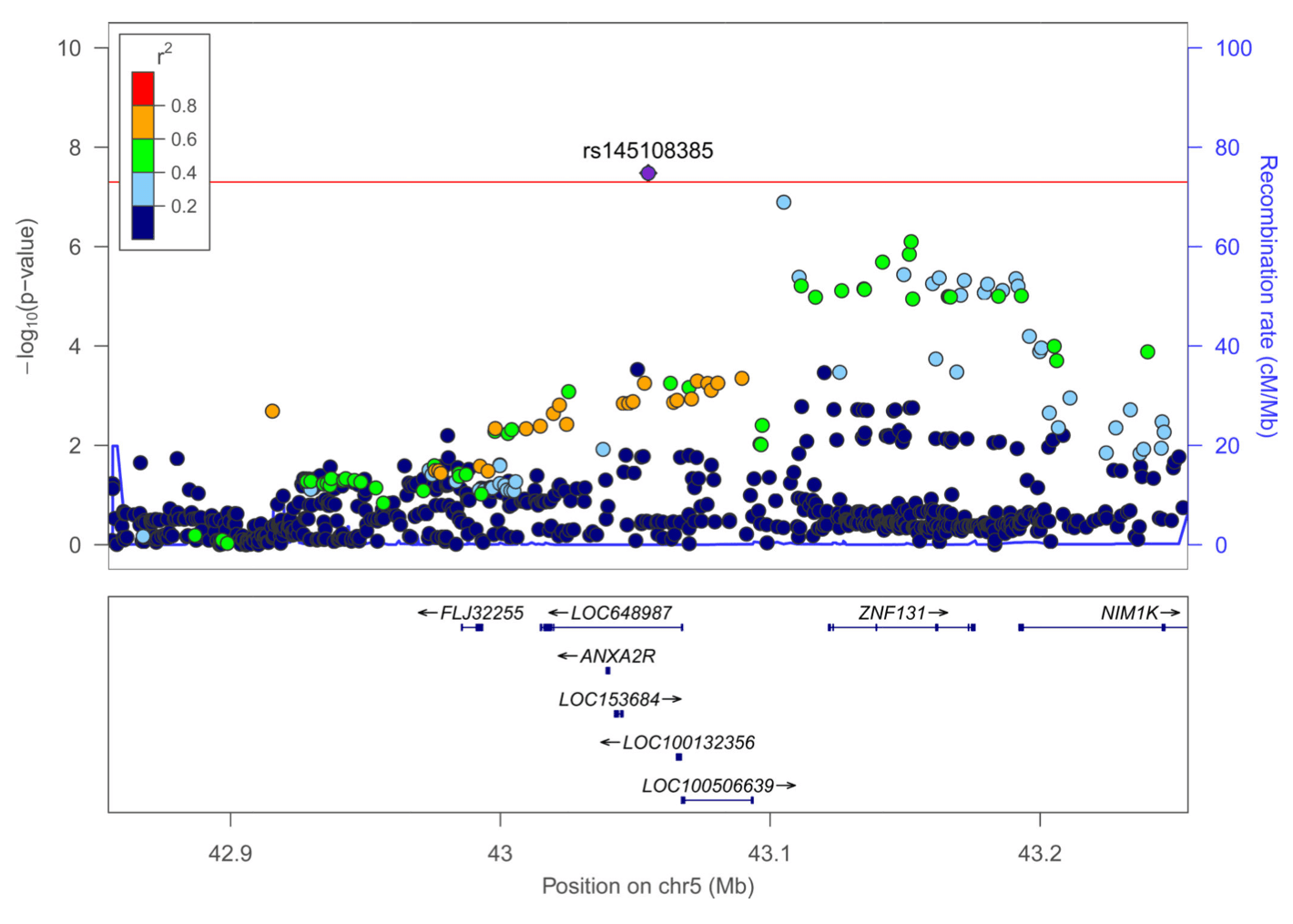
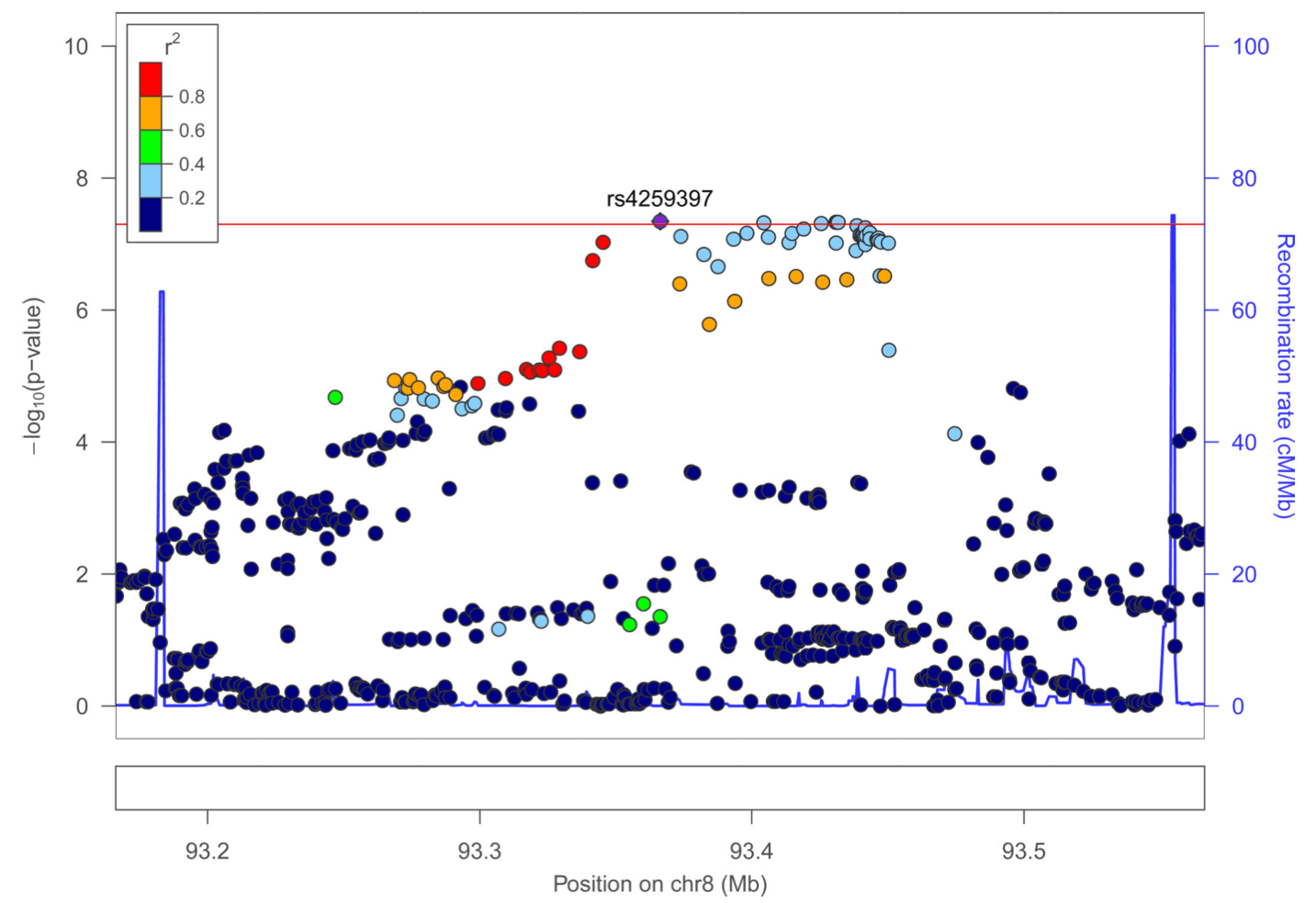
We undertook a fixed effects meta-analysis across ADHD and lifetime cannabis use GWAS results (Figure 1, Sup Figure 1) and obtained a genomic inflation factor of 1.22 (lambda 1000=1.006). This analysis found sixteen signals that met genome-wide significance (P<5×10−8) (Sup Table 2). Out of these, nine sentinel variants in seven regions did not meet genome-wide significance in the full PGC+iPSYCH ADHD or the cannabis use GWAS alone. Fixed effect and random effects meta-analysis results were consistent for these variants except for rs2391769 that showed evidence of heterogeneity between both studies (I=62.01, Sup Table 2). Seven of these variants were located in regions already reported by PGC+iPSYCH in the ADHD metaanalysis; conditional analyses showed that none of them were independent from the associations previously described by PGC+iPSYCH (Sup Table 3). The remaining signals, rs145108385 in chromosome 5 and rs4259397 in chromosome 8, lied in regions not formerly implicated by either PGC+iPSYCH or ICC meta-analyses (Figure 1, Figure 2). Rs145108385, with a P-value of 3.30×10−8 in the meta-analysis (full PGC+iPSYCH ADHD P=1.58×10−7 and cannabis use P=3.99×10−2) is an intronic SNP in LOC648987 and rs4259397, with a P-value of 4.52×10−8 in the meta-analysis (full PGC+iPSYCH ADHD P=3.68×10−6 and cannabis use P=6.20×10−3), is intergenic with the closest genes being FLJ46284 (+359 kb) and RUNX1T1 (−251 kb).
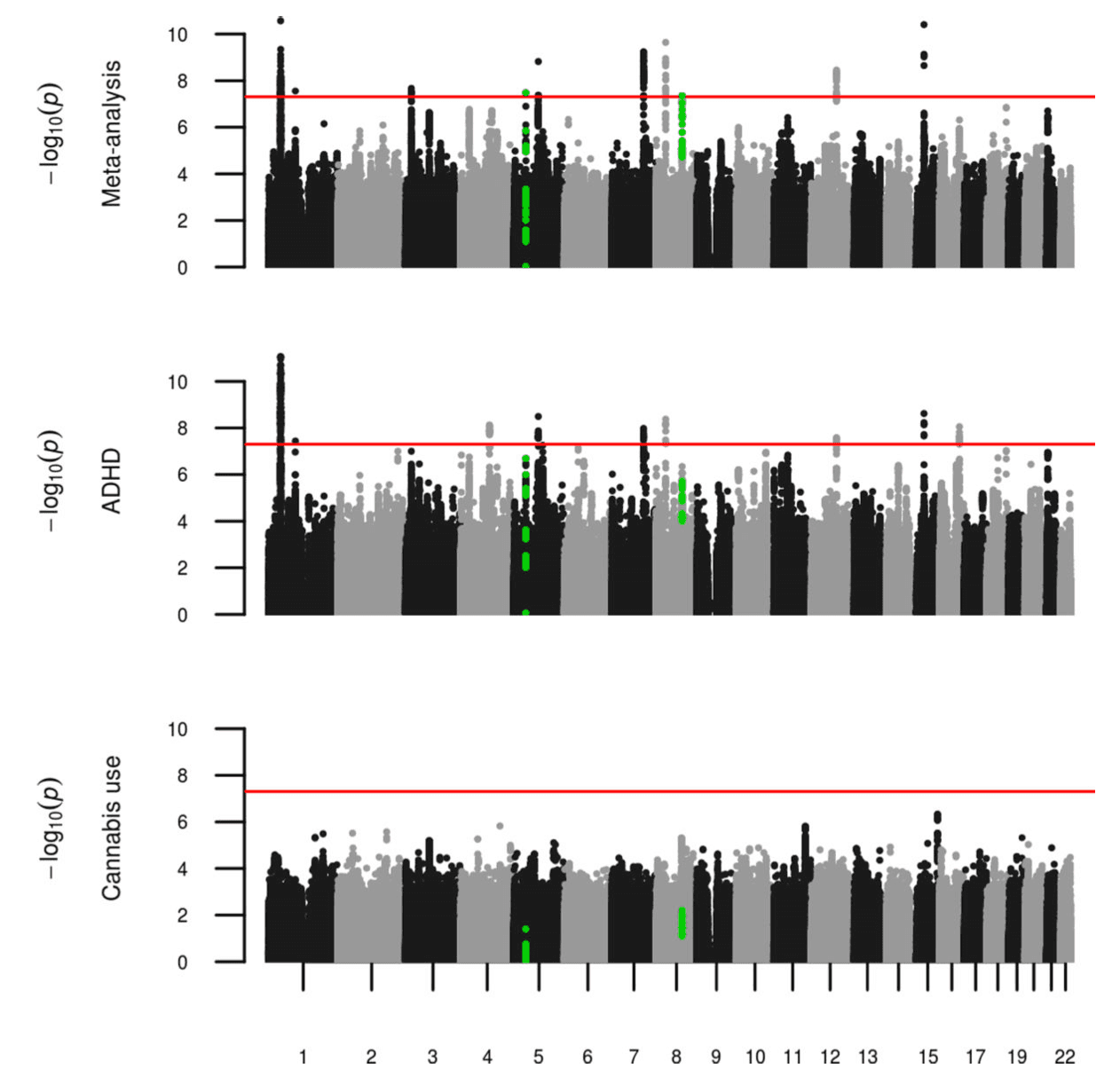
In the gene-based analysis, five genes, WDPCP, SLC9A9, TMEM161B, ZNF251 and ZNF517, met the Bonferroni corrected threshold for the number of genes analysed (P<2.79×10−6) in the cross-trait analysis but not in ADHD or cannabis use meta-analyses alone (Sup Table 4). Three of these genes also met the threshold in the random effects meta-analysis (WDPCP, TMEM161B and ZNF251, Sup Table 4). TMEM161B, however, lies in a locus identified for ADHD by the single variant analysis (rs4916723, Sup Table2).
Sign test
The sign test showed that variants associated with ADHD had a consistent direction of effect in the cannabis use analysis, with significant results for the following P-value thresholds: 5×10−6(P=6.72×10−3), 5×10−7 (P=3.50×10−2) and 5×10−8 (P=4.33×10−2) (Sup Table 5). Variants at none of the thresholds for cannabis use showed significant results in the sign test when testing the consistency of direction of effect in ADHD (Sup Table 5).
Mendelian randomization
The main analysis results for the most strict threshold for association with ADHD (P<5×10−8, 12 variants) showed evidence of a causal effect of ADHD on lifetime cannabis use (P=5.88×10−5, Table 1). The odds of cannabis use for ADHD versus non-ADHD indicate that individuals with ADHD were 7.9 times more likely to consume cannabis than those without ADHD (95 % CI (3.72, 15.51)). Sensitivity analyses showed consistent results overall (Figure 3), with the weighted median method being also significant (P=1.13×10−4, Table 1) and leave-one-out analyses providing evidence that this finding was not driven just by a single variant (Figure 3b)). Single variant results for ADHD and cannabis use, as well as IVW causal effect estimates for the 12 markers included in this analysis are provided in Sup Table 6. When using a more relaxed threshold for this comparison (P<5×10−6, 72 variants) results for all methods were weaker, although the main analysis remained significant (P=2.61×10−4, Table 1).
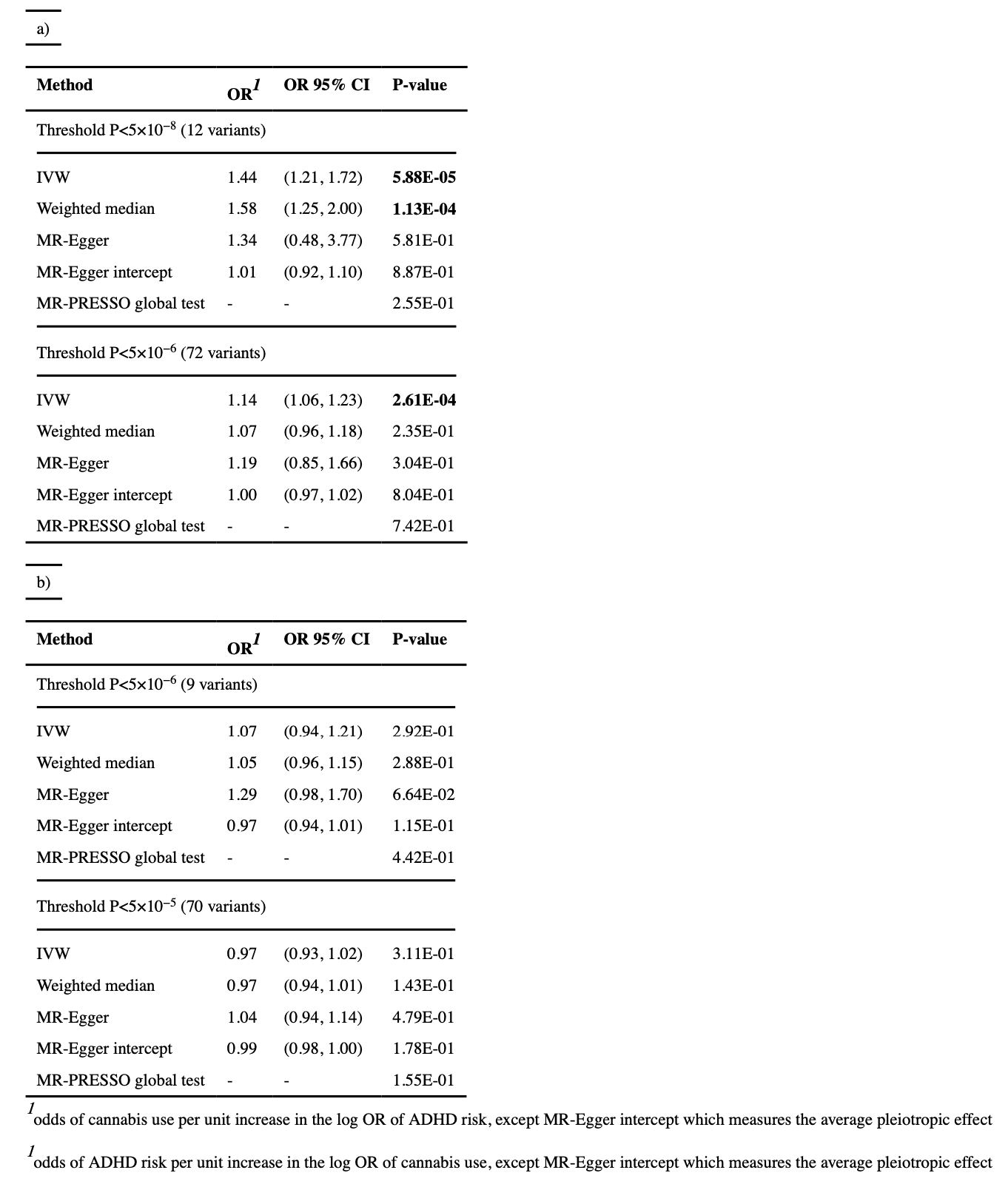
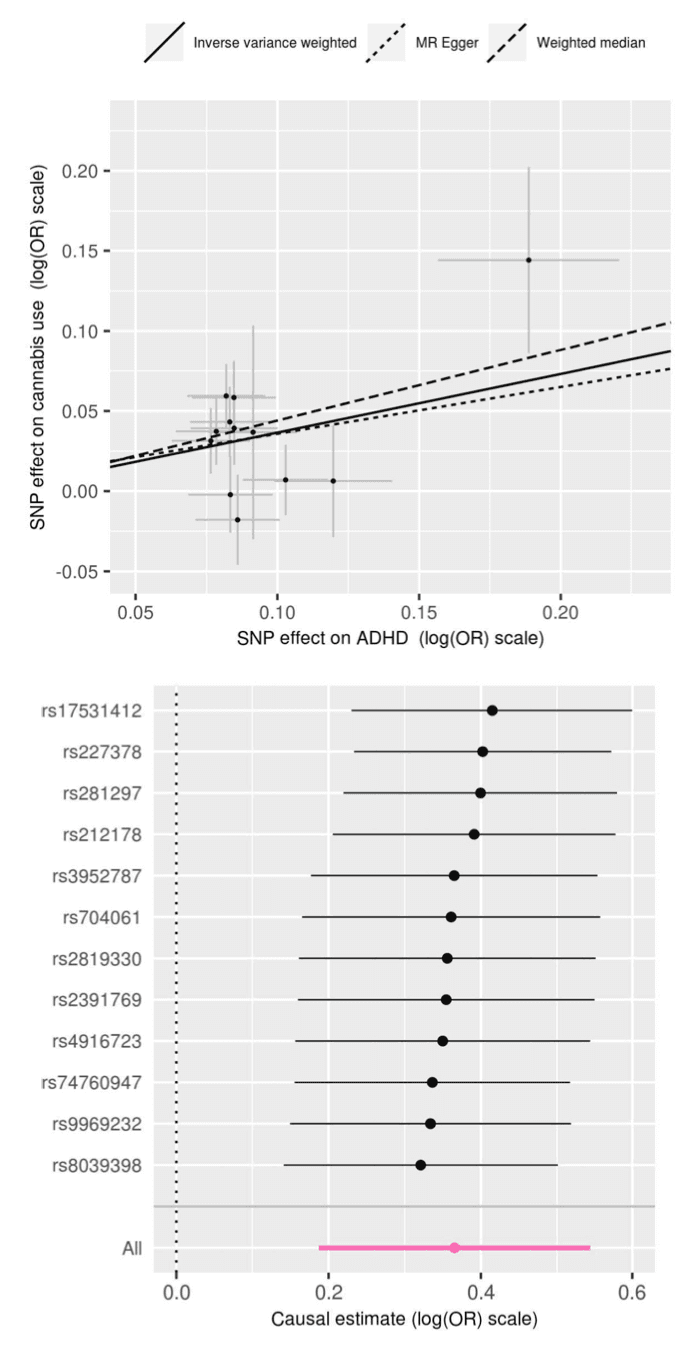
No evidence of a causal effect was detected with any of the thresholds or any of the methods for the association in the opposite direction (cannabis use as exposure and ADHD as outcome). No evidence of pleiotropy was found for any of the comparisons or any of the thresholds, either in the MR-Egger regression test of the intercept, MR-PRESSO global test (Table 1) or the heterogeneity test (Sup Table 7).
Discussion
To clarify the nature of the relationship between ADHD and cannabis use we estimated the genetic correlation between them, ran a cross-trait meta-analysis and inferred the causal role of ADHD on lifetime cannabis use by analysing current GWAS datasets of ADHD and cannabis use from the Psychiatric Genomic Consortium+iPSYCH and the International Cannabis Consortium.
In line with previous evidence supporting the co-occurrence of these two traits and the increased risk for cannabis use in individuals with ADHD, we found a highly significant genetic correlation between them. We also provided support for a causal link between ADHD and lifetime cannabis use.
These two conditions share a background of common genetic variants (rg=0.29, P=1.63×10−5) which may explain the phenotypic overlap observed between them and is consistent with previous genetic studies. However the genetic correlation alone does not distinguish between pleiotropy or causation. Strengthening the results of observational studies, the Mendelian randomization analysis provided significant evidence of a causal effect of ADHD on lifetime cannabis use. It estimated that individuals with ADHD are 7.9 times more likely to consume cannabis than individuals without an ADHD diagnosis. No support for the idea that cannabis use increases the risk of ADHD was found, which is consistent with prospective studies supporting that childhood ADHD is associated with cannabis use and cannabis disorder in adulthood. The sign test results also pointed to the same conclusion, showing that ADHD-associated variants had a consistent direction of effect on cannabis use.
To identify potential genetic mechanisms through which ADHD may increase the risk for cannabis use, we undertook a cross-trait analysis at SNP and gene levels, a powerful strategy to detect genetic variants with an effect in two or more genetically correlated traits.
This analysis identified four new genome-wide significant loci. The top hit of the SNP-based analysis, rs145108385, is an intronic variant at LOC648987 on chromosome 5. This variant earlier showed suggestive evidence of association (P=9 ×10−6) with squamous cell lung carcinoma and could point to a mechanism involved in smoking behavior. The second genome-wide significant hit, rs4259397, is an intergenic SNP on chromosome 8. It is located 251 kb upstream of RUNX1T1, which encodes a brain-expressed protein involved in transcriptional repression. Interestingly, an independent variant in the 3’ UTR of RUNX1T1, rs4500123, showed suggestive evidence (P=6 ×10−6) of association with oppositional defiant disorder in a GWAS of 750 ADHD cases from the International Multicentre ADHD Genetics (IMAGE). These results are in line with other studies indicating that oppositional behaviors in children are strong predictors for cannabis abuse and dependence, and highlight the importance of considering distinct patterns of co-occurrence of additional externalizing problems to strengthen the power of future genetic studies and to disentangle whether the association between ADHD and cannabis use is mediated by other externalizing behaviors.
At the gene level, we found evidence of genome-wide significant association for WDPCP and ZNF251. WDPCP encodes a cytoplasmatic WD40 repeat protein involved in the planar cell polarity signaling pathway and has been associated with major depression disorder and glucocorticoid receptor response. ZNF251 is highly expressed in fetal brain and cerebellum, lies in a duplication at 8q24.3 identified in sporadic autism spectrum disorder, and undergoes significant changes in its methylation status during fetal brain development. Despite not meeting the significance threshold in the random effects analysis, SLC9A9 is involved in synaptic transmission and plasticity, has been implicated in human ADHD and in rat studies of hyperactivity and has been found in multiple GWAS for addiction-related disorders.
In addition to the aforementioned loci, other interesting genes previously associated with ADHD in the meta-analysis run by the Psychiatric Genomics Consortium and iPSYCH remained statistically significant in the present cross-trait analysis. Genes such as FOXP2, PTPRF or SEMA6D, involved in neurodevelopmental processes, synaptic function, axon guidance or substance dependence and related phenotypes, may also be relevant in the risk for cannabis use. The involvement of genome-wide significant signals from the cross-trait analysis in the etiological links between ADHD and lifetime cannabis use deserves further investigation.
The results of the present study should be interpreted in the context of several methodological considerations:
First, Mendelian randomization uses genetic variants associated with an specific exposure to test whether this exposure causes an outcome. For this approach to be valid, certain assumptions need to be met, and a variety of methods exists to estimate the casual effect of the exposure on the outcome using genetic association summary statistics. In the present study we used the IVW method as the main analysis and the MR-Egger and weighted median as additional methods to assess the robustness of our results. We found no evidence of pleiotropy (which violates one of the assumptions) therefore the IVW estimate was preferred over the MR-Egger estimate, as it is more precise in the absence of pleiotropy. The strongest results were obtained when using a restrictive approach to select variants (P<5×10−8), and in this case the results were consistent when using the IVW and the weighted median methods. When using a more relaxed threshold (P<5×10−6), the effect estimates were reduced for all methods; the IVW result remained significant but the weighted median output did not. A possible explanation is that the association signal detected by the IVW method with the more relaxed threshold was still driven by the variants with stronger associations (mostly included in the more restrictive analysis); since the weighted median method uses the median of the ratio estimates (weighted by their standard error) of all variants, the signal is diluted when using this method. Relaxing the threshold potentially increases power by increasing the number of variants but, given that the strength of association for these variants is weaker, invalid instruments may also be introduced.
Second, no evidence of a causal effect was detected with any of the thresholds or any of the methods for the association of lifetime cannabis use as exposure and ADHD as outcome in the Mendelian randomization analysis. We cannot discard, from a purely statistical point of view, that this resulted due to lack of power, given the smaller sample size of the meta-analysis on lifetime cannabis use GWAS in comparison to the ADHD study, we may not have selected appropriate instruments to test the hypothesis that cannabis use increases the risk for ADHD.
Third, the causal effect estimate of cannabis use for ADHD versus non-ADHD presented here (OR=7.9, 95 % CI (3.72, 15.51)) needs to be interpreted with caution, given that winner’s curse bias may have lead to an inflation of the ADHD GWAS top results and this could have inflated the causal estimate. In addition, the limited number of variants included in this analysis contributes to the uncertainty of the estimate reflected by the wide confidence interval. Observational estimates of the effect of ADHD on lifetime cannabis use vary widely. A meta-analysis of prospective study estimates (8) provided an OR of 2.78 (95% CI (1.64, 4.74)) with study estimates ranging from 1.55 (95% CI (0.88, 2.72)) to 7.67 (95% CI (3.16, 18.64)) and a Cochran Q test of heterogeneity with a Q=20.38 and P<0.01. This heterogeneity may be affected by methodological variability, sample characteristics, follow-up length, study design (population based versus case-control) or assessment methods. Future Mendelian randomization studies using genetic effect estimates from a large number of robustly associated variants obtained in independent datasets, different from the discovery sets, will contribute to obtain more accurate causal estimates.
Forth, our cross-sectional study revealed a causal role of ADHD on cannabis use but gave no information about the relationship between ADHD symptoms, disorder presentations or other comorbid conditions and the risk for substance use. Given that specific ADHD symptom profiles and co-occurring disorders, including other externalizing disorders, influence substance use outcomes in ADHD clinical and population samples, their role in the causal effect of ADHD on lifetime cannabis use warrants further investigation. Considering cannabis use related outcomes, such as type, quantity, way of administration, or age at initial consumption of cannabis, may also help to clarify its relationship with ADHD.
In summary, we reported a genetic correlation between ADHD and lifetime cannabis use and provided support of a causal effect of ADHD on the risk for cannabis use through the analysis of genetic data. These results are in line with the temporal relationship between ADHD and future adverse health outcomes, reinforce the need to consider substance misuse in the context of ADHD in clinical intervention, and highlight the need for future genetic studies to provide insight into the shared biological mechanisms underlying both conditions.