Abstract
Human immunodeficiency virus (HIV) remains a persistent public health concern throughout the world. Substance use disorders (SUDs) are a common comorbidity that can worsen treatment outcomes for people living with HIV. The relationship between HIV infection and SUD outcomes is likely bidirectional, making clear interrogation of neurobehavioral outcomes challenging in clinical populations. Importantly, the mechanisms through which HIV and addictive drugs disrupt homeostatic immune and CNS function appear to be highly overlapping and synergistic within HIV-susceptible reward and motivation circuitry in the central nervous system. Decades of animal research have revealed invaluable insights into mechanisms underlying the pathophysiology SUDs and HIV, although translational studies examining comorbid SUDs and HIV are very limited due to the technical challenges of modeling HIV infection preclinically. In this review, we discuss preclinical animal models of HIV and highlight key pathophysiological characteristics of each model, with a particular emphasis on rodent models of HIV. We then review the implementation of these models in preclinical SUD research and identify key gaps in knowledge in the field. Finally, we discuss how cutting-edge behavioral neuroscience tools, which have revealed key insights into the neurobehavioral mechanisms of SUDs, can be applied to preclinical animal models of HIV to reveal potential, novel treatment avenues for comorbid HIV and SUDs. Here, we argue that future preclinical SUD research would benefit from incorporating comorbidities such as HIV into animal models and would facilitate the discovery of more refined, subpopulation-specific mechanisms and effective SUD prevention and treatment targets.
1. Introduction
Substance use disorders (SUDs) are chronic conditions characterized by enduring impairments in the control of motivated behavior that are often comorbid with other physical or neuropsychiatric disorders and diseases. Rates of human immunodeficiency virus (HIV) infection are much higher among individuals with SUDs compared to the general population, and SUDs are known to complicate HIV treatment efforts (Hartzler et al., 2017). For example, substance misuse is associated with reduced healthcare utilization and antiretroviral therapy (ART) adherence and with poor viral load management among people living with HIV (PLWH; Durvasula & Miller, 2014). Importantly, addictive substances may also impair ART efficacy through direct drug-drug interactions, which can contribute to less successful HIV treatment outcomes (Rasbach et al., 2013; Kumar et al., 2015). While PLWH can live long, relatively healthy lives with ART, many individuals who achieve viral suppression still develop HIV-associated neurocognitive disorder (HAND). Affecting nearly half of all PLWH (Heaton et al., 2010; Simioni et al., 2010), HAND is characterized by a spectrum of cognitive dysfunction, ranging from asymptomatic neurocognitive impairment to HIV-associated dementia (Clifford and Ances, 2013), although the vast majority of HAND cases in the current ART era are asymptomatic and only identified via cognitive testing (Vastag et al., 2022). HIV may also exacerbate drug-induced cognitive dysfunction and age-related cognitive decline (Becker et al., 2004; Valcour et al., 2004a; Norman et al., 2009; Alford and Vera, 2018), which complicates long-term health outcomes for individuals living with SUDs. These concerning realities highlight the need for targeted therapeutics to treat SUDs in PLWH. The preclinical application of translational animal models is vital towards this goal.
Since the late 1980s, research has indicated that use of addictive drugs is inextricably linked to increased HIV risk (Weiss, 1989), which is most commonly due to increased risk-taking behaviors such as needle sharing and unprotected sex. Preclinical animal models have been instrumental towards improving our understanding of the neurobiological and behavioral sequelae of comorbid HIV and SUDs. We discuss several prominent animal models of HIV and describe key findings that characterize the pathophysiological milieu of these models. We then highlight key preclinical findings across numerous animal models of comorbid HIV and SUDs and identify lingering gaps in the literature that require further research. Finally, we provide selected examples of modern, cutting-edge behavioral neuroscience tools within these preclinical models of HIV and SUDs that we expect to advance our knowledge of the neurobiological and behavioral intersections of comorbid HIV and SUDs. As highlighted in Table 1, HIV animal models come with unique caveats that ultimately constrain the interpretations and extrapolations one can generate from these models. Nevertheless, we broadly argue that future medications development efforts to address SUDs must consider comorbidities such as HIV to improve treatment outcomes and that next-generation neuroscience tools will aid in revealing novel therapeutic targets.
2. Animal Models of NeuroHIV
A broad spectrum of rodent and non-human primate (NHP) animal models has been developed to understand the pathophysiology of HIV and its impact on central nervous system (CNS) function. Models ranging from exogenous HIV-1 protein exposure to HIV-1 transgenic rodents and HIV-like viral infections in NHPs have provided crucial insights into the neurocognitive and behavioral outcomes of HIV, many of which parallel clinical observations of cognitive and behavioral impairments among PLWH (Jaeger and Nath, 2012; Clifford and Ances, 2013; Moran et al., 2014; Mallard and Williams, 2018). Importantly, research from animal models of neuroHIV indicates that mesocorticolimbic reward circuits are particularly vulnerable to HIV and its protein products. Further, convergent research indicates that HIV interacts additively or synergistically with addictive drugs to promote reward system and neuroimmune dysfunction and neurocognitive impairment.
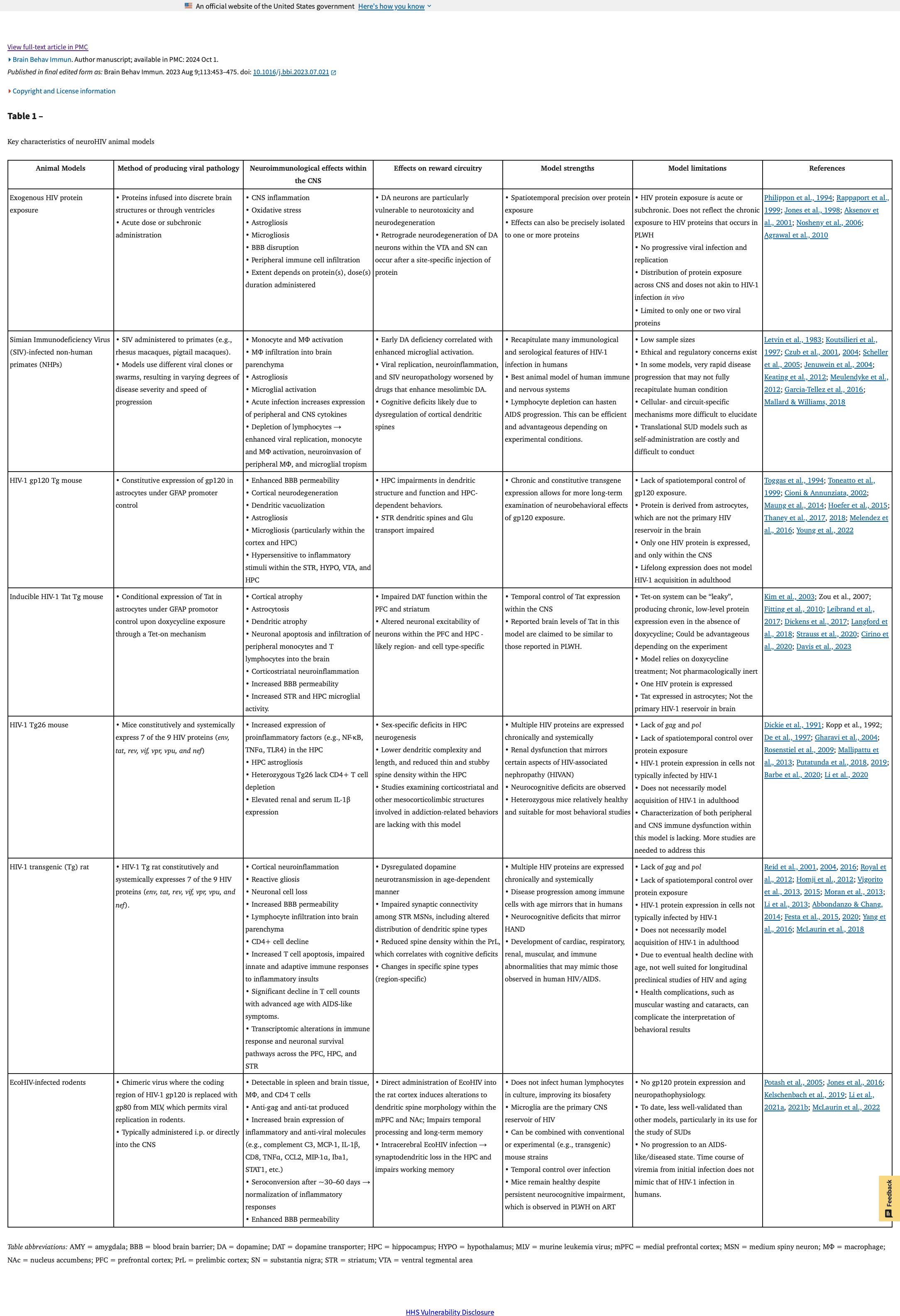
HIV enters the CNS predominantly via infected monocytes and T cells, where it establishes a productive infection within microglia (Figure 1). Within the periphery and the brain parenchyma, HIV-induced dysregulation of immune signaling, such as through upregulation or suppression of various cytokines and chemokines, can disrupt homeostatic neuronal function, leading to impaired cognition and behavior. PLWH exhibit a persistent viral reservoir within the CNS that contributes to neuroHIV and HAND. This persistence of chronic, low-levels of HIV protein within the CNS may contribute to HIV-associated neuroimmune, cognitive, and behavioral dysfunction. Preclinical animal models each capture a unique set of key pathophysiological features of neuroHIV and HAND and vary in key immunological characteristics captured in the model, each with distinct advantages and disadvantages (Table 1).
2.1. Exogenous HIV Protein Exposure
One in vivo methodology to study the neurobiological and behavioral sequelae of neuroHIV is the administration of HIV proteins, such as envelope glycoprotein 120 (gp120) and trans-activator of transcription (Tat), directly into the CNS. Both systemic and neuroanatomically discrete administration of HIV proteins have been examined across numerous studies. Early in vitro studies provided clear evidence of HIV protein-induced neurotoxicity (Lipton, 1991; Sabatier et al., 1991; Müller et al., 1992; Bennett et al., 1995; Weeks et al., 1995; Yeung et al., 1995; Nath et al., 1996), which served as the foundation for in vivo experiments in rodents probing the neurotoxic effects of HIV protein exposure within mesocorticolimbic reward circuitry. One of the first studies to examine HIV protein neurotoxicity in vivo found that microinfusions of the basic domain of the Tat peptide into the brain – including administration to the lateral ventricles, hippocampus, or thalamus - of mice produced neuroinflammation and reactive astrogliosis. These deficits were attenuated by pharmacological inhibition of the proinflammatory cytokine tumor necrosis factor alpha (TNFα) (Philippon et al., 1994), implicating proinflammatory signaling as critical mediator of Tat-induced CNS dysfunction (Rappaport et al., 1999).
Intracerebroventricular (i.c.v.) administration of HIV-1 Tat in rats is also associated with increased astrocytosis and peripheral immune cell infiltration into the parenchyma (Jones et al., 1998), suggesting impaired blood brain barrier (BBB) permeability induced by HIV protein exposure. In addition to Tat effects on the BBB, HIV-1 gp120 administration into the dorsal striatum of rats enhances the activation of extracellular matrix metalloproteinase 2 (MMP-2) and MMP-9 and downregulates essential proteins for regulation of BBB integrity – laminin and claudin-5 –, which is consistent with impaired BBB integrity (Louboutin et al., 2010). Interestingly, striatal MMP-2 and MMP-9 activity have been implicated in drug-seeking behavior (Smith et al., 2014; Namba et al., 2022), while changes in claudin-5 expression and reduced BBB integrity are associated with depression-like behaviors (Menard et al., 2017). Other studies show that mice exposed to Tat exhibit increased neuroinflammation within the CNS and depression-like behavior (Pu et al., 2003; Lawson et al., 2011). Viral vector-mediated overexpression of antioxidants such as glutathione peroxidase and Cu/Zn superoxide dismutase can attenuate HIV protein-induced oxidative stress (Louboutin et al., 2010; Agrawal et al., 2012). Taken together, these studies provide clear evidence of neuroinflammatory and neurotoxic consequences of direct administration of HIV proteins into the CNS, and further studies have demonstrated mesocorticolimbic circuit dysfunction following HIV protein exposure.
Exogenous HIV protein administration into the CNS elicits neurodegenerative and neurotoxic effects on dopamine neurons (Nath et al., 2016; Gaskill et al., 2017). For example, direct administration of HIV-1 Tat into the rat striatum – a primary target of midbrain dopamine projections – reduced tyrosine hydroxylase (TH) staining within dopamine cell bodies in the substantia nigra (SN) and caused Parkinson’s-like locomotor deficits, thus implicating dopamine projection neurons as vulnerable to HIV protein exposure (Zauli et al., 2000). Microinfusions of Tat or gp120 within the rat striatum increased cell death, induced reactive astrogliosis, and enhanced oxidative stress (Bansal et al., 2000; Aksenov et al., 2001, 2003). Further investigation into the mechanism by which HIV proteins induce these neurotoxic effects revealed supporting evidence of retrograde neurodegeneration of dopamine neurons induced by intra-striatal gp120 administration (Nosheny et al., 2006), an effect that is attenuated by viral vector-mediated overexpression of brain-derived neurotrophic factor (BDNF; Mocchetti et al., 2007). In support of the hypothesis that dopamine neurons are particularly vulnerable to the neurotoxic effects of HIV, dopamine neurons of rats exposed to intra-striatal gp120 exhibit more rapid degeneration, and at lower gp120 doses, than non-dopamine neurons. These effects were prevented by viral-vector mediated overexpression of antioxidant enzymes (Agrawal et al., 2010). Altogether, these studies highlight the complex neuroinflammatory and neurotoxic effects of HIV protein exposure within the CNS and implicate mesocorticolimbic reward circuitry as particularly vulnerable to HIV infection.
One primary advantage of the protein exposure models is their spatiotemporal precision. Specifically, these models allow investigators to expose discrete brain regions to HIV proteins, at specific doses and experimental timepoints of interest, which can be particularly useful when assessing the interactions between HIV and addictive substances on cognition and behavior. However, there is a paucity of studies that directly compare the impact of systemic (e.g., i.c.v.) versus neuroanatomically specific administration of HIV proteins on neurotoxicity, neuroinflammation, and impairments in neuronal physiology and behavior, which is particularly important given the potential for local exposure to HIV proteins to have broad neuroinflammatory effects. Another distinct advantage of these models, which complements their spatiotemporal precision, is that they allow isolation of HIV-associated pathophysiological processes to one or more specific HIV proteins. One key limitation of these protein exposure approaches is that they are generally limited to acute or subchronic exposure periods and to only one or two viral proteins, which does not necessarily reflect the chronic, persistent exposure to multiple HIV proteins that occurs in PLWH. The doses used in many studies are also often much higher than the expected concentration of HIV protein within the CNS of PLWH, and the distribution of viral protein throughout the CNS may not reflect what is observed in humans. Further, there is no progressive viral infection and replication, which limits the translational extrapolations one can draw from these protein exposure studies. As described below, other animal models have been developed to address these caveats.
2.2. SIV-infected Non-Human Primates (NHPs)
Immunodeficiency in captive macaque monkeys that resembled human acquired immunodeficiency syndrome (AIDS) was first reported in 1983 by Letvin and colleagues (Letvin et al., 1983), and the cause of this condition was eventually identified as Simian Immunodeficiency Virus (SIV; Bailes et al., 2003; Gao et al., 1999; Peeters et al., 1989). HIV-1 is closely related to SIVcpz, which infects chimpanzees of West-Central Africa and is believed to be the origin of the HIV-1 group ‘M’ that comprises the majority of HIV-1 infections in humans (Simon et al., 1998; Nerrienet et al., 2005). Similarly, HIV-2 is closely related to SIVsmm, which infects sooty mangabeys (Lemey et al., 2003). Infecting macaques with HIV-1 is difficult due to cellular proteins found in macaques that restrict HIV-1 replication (Stremlau et al., 2004; Misra et al., 2013). Today, many NHP models of HIV infection utilize rhesus macaques infected with various strains of SIVmac, which recapitulates many immunological and serological features of HIV-1 infection in humans (Garcia-Tellez et al., 2016). Considered the ‘gold-standard’ animal model of HIV infection in humans (Mallard and Williams, 2018), SIV-infected NHPs have been crucial to our understanding of the pathophysiology of neuroHIV.
Akin to HIV protein administration into the CNS of rodents, SIV infection in NHPs produces profound neuroimmune dysfunction. Early work investigating neuroinvasion of SIV in rhesus monkeys found that SIV facilitates macrophage infiltration into the brain parenchyma as early as 7 days post-inoculation, which coincides with enhanced microglial activation and neurovascular injury (Chakrabarti et al., 1991). More recently, studies have utilized CD4 or CD8 T lymphocyte depletion to enhance viral replication in macrophages, neuroinvasion of infected monocytes/macrophages into the brain parenchyma, and tropism for microglia. For example, depletion of CD8 lymphocytes and infection with the viral swarm SIVmac251 results in rapid onset of AIDS and a higher rate of SIV encephalitis (Williams et al., 2005). Similarly, CD4 lymphocyte depletion prior to SIV infection also results in enhanced viral replication and progression to AIDS as well as increased microglial infection (Micci et al., 2014). Circulating cytokine and chemokine levels within the CNS and the periphery of SIV-infected macaques are also dysregulated (Keating et al., 2012). Acute infection with SIVmac251 results in a transient upregulation of IL-1β, IL-6, IL-10, and TNFα mRNA expression in peripheral blood mononuclear cells (PBMCs), which may produce early disruptions to BBB integrity and contribute to early CNS infection (Benveniste et al., 1996). A recent study demonstrated that SIVmac251 infection upregulates CCL2, IL-6, CXCL10, and IFNγ levels within the cerebrospinal fluid, and ART treatment only significantly attenuates the increase in CXCL10 (Solis-Leal et al., 2022). Consistent with these findings, ART treatment does not completely resolve HIV-induced dysregulations to peripheral cytokine expression in PLWH (Keating et al., 2011), which may be a mechanism underlying the persistence of HIV within the CNS as well as HAND in the ART era. These studies collectively highlight the importance of peripheral immune cell activity and CNS neuroimmune signaling as key mechanisms that mediate HIV-induced neuropathogenesis.
SIV-infected rhesus macaques also exhibit both motor and cognitive deficits that are not specifically related to the extent and anatomical location of virus-induced lesions within the brain (Murray et al., 1992). Moreover, reactive astrogliosis in SIV-infected macaques is associated with the onset and progression of neuropsychological impairments regardless of immunodeficiency syndrome (Rausch et al., 1994). A time-dependent increase in reactive astrogliosis and synaptic impairments was observed within the frontal cortex of SIV-infected macaques, which is a likely mechanism mediating the cognitive deficits observed in the previous studies (González et al., 2000). Another early study demonstrated that SIVmac251-infected macaques exhibit dendritic spine loss as early as 2.5–3 months post-infection (Montgomery et al., 1999), suggesting that early CNS infection may impair cortical synapses and promote cognitive dysfunction. These studies provided early evidence for the hypothesis that SIV and HIV can rapidly invade the CNS and elicit significant neurological, cognitive, and behavioral deficits through more global CNS dysregulation irrespective of virus-induced neurotoxicity or disease severity. These findings also suggest that high concentrations of HIV protein within a discrete brain region may not be necessary to produce impairments within that region, which helps inform lingering gaps in the literature regarding the differences between local versus systemic exposure to exogenous HIV protein. As SIV-infected macaques treated with ART still have detectable SIV env DNA within the basal ganglia and brain stem, CNS viral reservoirs may persist within the mesocorticolimbic system even with ART treatment (Perez et al., 2018). The chronic, low-level HIV proteins secreted by these viral reservoirs may contribute to global CNS dysregulation. This has significant implications for the present-day clinical landscape of neuroHIV in humans, where chronic neuroinflammation and CNS reservoirs of HIV can persist despite undetectable viral loads in the periphery (Heaton et al., 2010; Sari et al., 2022).
Beyond the direct contribution to our understanding of the preclinical landscape of neuroHIV and SUDs, many studies using the SIV model to study HAND have revealed important insights into HIV-induced neurocognitive dysfunction that could have important implications for the study of comorbid HIV and SUDs. Early studies in NHPs demonstrated that SIV infection in rhesus macaques can produce psychomotor impairments (e.g., slower choice reaction times and forelimb movement) as well as cognitive and behavioral dysfunction that includes impaired discrimination learning, memory retention, attention, and motivation (Murray et al., 1992; Gold et al., 1998; Marcario et al., 1999; Weed et al., 2003). These SIV-induced behavioral impairments do not appear to be dependent on discrete virus-induced lesions within the brain (Murray et al., 1992). Indeed, neuroimmune activation and subsequent neuronal dysfunction may underlie these psychomotor and cognitive impairments. For example, one study demonstrated a significant correlation between psychomotor impairments in SIV-infected macaques and associated axonal damage, microglial activation, and peripheral macrophage infiltration (Weed et al., 2003). Exposure to addictive substances may exacerbate these neurobehavioral effects. For example, SIV-infected NHPs injected with methamphetamine exhibit increased CCR5 expression in the brain, which is correlated with viral load, and methamphetamine exposure also increases the proportion of microglia infected by SIV within the CNS (Najera et al., 2016; Niu et al., 2020). Opioids such as morphine may also augment the pathophysiology of SIV infection and enhance virus-induced neurocognitive deficits (Reddy et al., 2012; Marcario et al., 2016; Acharya et al., 2021). However, these effects likely depend on drug type, duration of drug exposure, and withdrawal (Molina et al., 2011; Weed et al., 2012; Wang et al., 2019b). Nevertheless, most studies probing the interactions between SIV infection and addictive substances utilize experimenter-delivered methods of drug exposure, which does not model how humans consume drugs. Drug self-administration models (Section 3) will advance study of the impact of drug use on HAND-like cognitive impairment in the SIV model, although there are additional limitations to consider here (Table 1).
Like rodents exposed to exogenous administration of HIV proteins within the CNS, SIV-infected NHPs display impairments in mesocorticolimbic monoamine transmission. SIV-infected macaques exhibit elevated levels of the dopamine metabolite 3,4-dihydroxy-phenylacetic acid (DOPAC) within the cerebrospinal fluid (CSF), which is also accompanied by a progressive, time-dependent increase in serotonin metabolites within 8 months from initial infection (Koutsilieri et al., 1997). While administration of selegiline (a monoamine oxidase inhibitor that increases CNS dopamine availability) or levodopa (L-DOPA) to SIV-infected macaques restored SIV-induced dopamine deficiency, it also worsened CNS viral replication and SIV-induced neuropathology (Czub et al., 2001). Both selegiline and L-DOPA also potentiate proinflammatory TNFα mRNA expression across the mesocorticolimbic reward system in SIV-infected macaques (Czub et al., 2004, although see Emanuel et al., 2022). Reduced dopamine levels within the striatum of SIV-infected macaques correlate with increased microglial activation (Scheller et al., 2005), and treatment with minocycline – which blocks microglial activation - can ameliorate this striatal dopamine decline (Meulendyke et al., 2012). This perhaps suggests that aberrant microglial activation promotes mesolimbic dopamine deficiency and the potential for bidirectional interaction between neuroimmune and monoamine outcomes. However, one study observed dopamine deficiency within the nucleus accumbens (NAc) of asymptomatic, SIV-infected macaques prior to any HIV-induced neuropathology, indicating that impaired monoamine signaling may precede HIV-induced neuropathogenesis (Jenuwein et al., 2004). These studies collectively suggest that HIV-induced monoamine impairments could contribute to HIV-associated neuroinflammation and neurocognitive dysfunction and that dopamine-modulating drugs, such as addictive drugs, may exacerbate these processes. Akin to findings from exogenous HIV protein models described above, these systems represent common mechanisms of HIV-induced pathology within the CNS that could be effective treatment targets. Despite the tremendous advancements produced by SIV-infected NHP models of HIV, work with NHPs comes with certain limitations that include low sample sizes as well as ethical and regulatory concerns (J. D. Estes et al., 2018; Table 1). Moreover, another limitation of NHP models is that there are generally fewer validated tools, compared to rodent models, that can precisely elucidate molecular, cellular, and circuit-specific mechanisms that mediate neurobehavioral impairments produced by combined HIV and drug use. The advent of sophisticated transgenic rodent models and chimeric HIV viruses that infect murine cells have greatly facilitated the investigation of the neural underpinnings of comorbid HIV and SUDs.
2.3. Transgenic Mice
Several transgenic (Tg) mouse models that express HIV-1 proteins, either constitutively or conditionally, have been utilized to model some aspects of the pathophysiology produced by HIV-1 infection within the CNS. One such model is the HIV-1 gp120 Tg mouse, which constitutively expresses gp120 in astrocytes under the control of the glial fibrillary acidic protein (GFAP) promoter. First described by Toggas et al., these mice exhibit cortical neurodegeneration, dendritic vacuolization and synapse loss, astrogliosis, and microglial activation across the brain (Toggas et al., 1994; Kang et al., 2010; Maung et al., 2014; Thaney et al., 2017, 2018). These mice also exhibit impaired long-term potentiation (LTP) within the CA1 region of the hippocampus, which likely contributes to spatial working memory deficits seen in these mice (Krucker et al., 1998; Hoefer et al., 2015). As in SIV-infected NHP and exogenous protein models, HIV-1 gp120 Tg mice also exhibit enhanced BBB permeability (Toneatto et al., 1999; Cioni and Annunziata, 2002; Strazza et al., 2011), suggesting that chronic gp120 exposure may facilitate HIV-1 neuroinvasion. A recent in vivoPET imaging study revealed an increase in translocator protein (TSPO) binding, which is indicative of microglial activation in mice, in multiple neural substrates (the striatum, hypothalamus, ventral tegmental area (VTA), and hippocampus) of Tg mice in response to lipopolysaccharide (LPS) exposure, suggesting a hypersensitive response to inflammatory stimuli within the mesocorticolimbic reward system of these mice (Young et al., 2022). Indeed, this has significant implications for SUDs, where PLWH may experience greater neuroinflammation and subsequent neurocognitive impairment due to the proinflammatory effects of many addictive drugs (Cui et al., 2014; Namba et al., 2021).
One disadvantage of the constitutive HIV-1 gp120 Tg mouse model is the lack of temporal control over CNS exposure to HIV proteins. To overcome this barrier, a conditional Tg mouse line that expresses HIV-1 Tat in GFAP+ cells upon doxycycline (i.e., a tetracycline antibiotic) exposure was developed. This mouse model produces Tat protein in astrocytes in a doxycycline-dependent manner that can be released to directly interact with neurons (for review, see Langford et al., 2018). This results in cortical atrophy, astrocytosis, dendritic degeneration, neuronal apoptosis, and infiltration of peripheral monocytes and T lymphocytes into the brain parenchyma (Kim et al., 2003a). These mice also exhibit corticostriatal neuroinflammation, increased BBB permeability, and increased striatal and hippocampal microglial activity (Kim et al., 2003a; Fitting et al., 2010; Leibrand et al., 2017). In addition to immune dysregulation, HIV-1 Tat Tg mice have altered dopamine transmission within the PFC and striatum. For example, Tat induction in these mice increases phasic dopamine release within the dorsal striatum through dopamine transporter (DAT) inhibition and stimulation of synaptic release of dopamine (Davis et al., 2023). Another study demonstrated that within the PFC, Tat induction reduces DAT-mediated dopamine reuptake and concomitantly inhibits action potential firing among layer V prelimbic cortex pyramidal neurons (Strauss et al., 2020). In contrast, another study showed increased action potential firing in layers II/III of the mPFC and decreased firing within the CA1 region of the hippocampus (Cirino et al., 2020). Given the significant immunomodulatory role of dopamine (Gaskill et al., 2017; Nolan and Gaskill, 2019; Xia et al., 2019), this dysregulation of dopamine homeostasis within the reward system could further impair neuroimmune signaling, thus contributing to Tat-induced neuropathology. However, as these studies highlight, it is possible that Tat-mediated dopamine impairments and subsequent dysfunction of neuronal activity could be brain region-specific.
While this model provides experimenters with greater temporal specificity over HIV protein expression, the expression of Tat in astrocytes represents a limitation of this model given that HIV primarily infects microglia in the CNS. Moreover, these tetracycline-inducible transgene expression systems are “leaky” and can produce chronic, low-level protein HIV-1 protein expression even in the absence of doxycycline (Fitting et al., 2010). Depending on perspective and experimental question, this could be viewed as either an advantage or disadvantage. On the one hand, uncontrolled expression of Tat detracts from the temporal control feature of this model. However, this may better model the chronic, low-level HIV protein exposure that occurs with neuroHIV among virally-suppressed individuals. In support of this, a recent study demonstrated that mice chronically exposed to low-level Tat (in the absence of doxycycline treatment) exhibit decreased cortical expression of the synaptic markers synaptophysin and PSD95 as well as increased hippocampal astrocytosis and neuroinflammation (Dickens et al., 2017). Constitutive, systemic expression of Tat in Tg mice is associated with an increase in evoked cortical glutamate release and a concomitant decrease in GABA release (Zucchini et al., 2013), suggesting that mice chronically exposed to HIV-1 Tat may exhibit impairments in cortical excitatory neurotransmission, which has important implications for the pathophysiology of SUDs (Kalivas, 2009; Koob and Volkow, 2016). It is important to note that doxycycline treatment is typically administered for shorter time periods (e.g., about one week) via i.p. injections or for longer periods (e.g., several weeks) orally via doxycycline-containing chow, and these different methods of administration can produce differential effects on neuropathology and behavior. Specifically, anxiety-like behavior, motor function, as well as spatial memory and reversal learning exhibit differential profiles of impairment between these two exposure paradigms (Joshi et al., 2020). Moreover, the neuropathology produced in the brain, such as gliosis, dysregulation of neuroimmune signaling, and impaired neurotransmission, differs between i.p. doxycycline-treated mice compared to doxycycline chow-fed mice (Kim et al., 2003b; Bruce-Keller et al., 2008; Fitting et al., 2010; Carey et al., 2012, 2013; Miller et al., 2018), emphasizing the need to consider the duration and route of doxycycline administration in inducible Tg models.
One limitation of the HIV Tg mouse models discussed above is the lack of chronic, systemic expression of multiple HIV proteins. This is resolved in the HIV-1 Tg26 mouse model, where a replication-incompetent HIV-1 provirus lacking gag and pol expresses the other seven HIV-1 genes (Dickie et al., 1991). These mice express high levels of HIV-1 transcripts env, tat, rev, vif, vpr, vpu, and nef in various tissues, such as skin, skeletal muscle, and brain. The homozygous Tg26 mice exhibit psoriasis-like skin lesions and progressive renal disease, while the heterozygous Tg26 mice have a longer lifespan and develop renal disease later in life (Dickie et al., 1991; De et al., 1997; Rosenstiel et al., 2009). In particular, Tg26 mice backcrossed with the C57BL/6 strain exhibit longer lifespans with fewer health complications (e.g., minimal renal disease; Gharavi et al., 2004; Mallipattu et al., 2013; Zhong et al., 2005), making these mice a more reliable model for studying HAND. These Tg26 mice also exhibit lower levels of viral transcripts within the brain, perhaps resembling PLWH on ART, which contributes to the translational potential of this model (Putatunda et al., 2018). Furthermore, studies on Tg26 mice have revealed deficits in hippocampal dendritic morphology and sex-specific spatial learning impairments (Putatunda et al., 2018, 2019; Barbe et al., 2020). A recent study demonstrated increased neuroinflammation and astrogliosis within the hippocampus of Tg26 mice (Li et al., 2020), which could potentially contribute to these behavioral and synaptic impairments. These findings point to the potential value of future studies examining addiction-related behaviors within this model.
2.4. HIV-1 Transgenic Rat
Akin to HIV-1 Tg26 mice, the HIV-1 Tg rat constitutively and systemically expresses 7 of the 9 HIV proteins and exhibits many pathological features of HIV infection in humans (Reid et al., 2001; Vigorito et al., 2015). These rats were first reported to exhibit many clinical aspects of AIDS by 5–9 months of age, including cataracts, wasting, respiratory difficulty, neurological abnormalities, and skin lesions (Reid et al., 2001). However, later studies using Tg rats obtained from a commercial vendor observed these symptoms at 18 months or older (Moran et al., 2013; Vigorito et al., 2013). Akin to observations in humans, CD4+ and CD8+ cells from HIV-1 Tg rats exhibit increased susceptibility to activation-induced apoptosis and diminished capacity to generate IFNγ following activation (Reid et al., 2004). Upon exposure to an inflammatory stimulus such as LPS, HIV-1 Tg rats also exhibit an exaggerated cytokine and chemokine response within the spleen and brain compared to controls, suggesting that non-replicative HIV-1 provirus expression in these rats may prime the immune system to produce dysregulated responses to future insults (Homji et al., 2012a). These rats also exhibit a decline in T cells beginning by around 6 months, which is exacerbated with advanced age alongside increased expression of proinflammatory factors such as IL-6 and TNFα (Abbondanzo and Chang, 2014). Overall, the immune system changes that occur over the lifespan of these rats mirrors, to an extent, what is observed in humans. However, the marked health decline of these animals with advanced age should be noted when designing studies with this model, particularly in the case of longitudinal studies and those pertaining to aging and HIV. Moreover, the constitutive expression of viral proteins in cell types not normally infected by HIV-1, which occurs in this model, represents a limitation that should be considered when interpreting findings from this model.
Neuropathology in the HIV-1 Tg rat consists of cortical neuroinflammation, reactive gliosis, neuronal cell loss, and increased BBB permeability and lymphocyte infiltration into the brain parenchyma (Reid et al., 2001; Royal et al., 2012). Within the striatum, HIV-1 Tg rats exhibit loss of TH expression that worsens with age, suggesting a time-dependent dysregulation of striatal dopamine neurotransmission (Reid et al., 2016). Importantly, these animals exhibit significant impairments in synaptic connectivity within the striatum, which includes profound alterations in the distribution of dendritic spine types on medium spiny neurons (MSNs; McLaurin et al., 2018). Specifically, HIV-1 Tg rats exhibit an increase in thin and mushroom spines proximal to MSN somas and an increase in stubby spines on more distal dendritic branches, as well as sex-dependent morphological alterations in dendritic complexity and organization (McLaurin et al., 2018). Transcriptome sequencing of the prefrontal cortex (PFC), hippocampus, and striatum from HIV-1 Tg rats revealed significant alterations in immune response and neuronal survival pathways across all three brain regions (Li et al., 2013), which may contribute to enduring changes in mesocorticolimbic synaptic connectivity and dendritic spine morphology. Significant alterations in cytokine and chemokine expression are observed in the PFC, NAc, and VTA of HIV-1 Tg rats compared to controls as well as differential immune responses to nicotine treatment (Yang et al., 2016). Within the PFC specifically, these rats exhibit reduced dendritic spine density that is likely driven by gp120-induced upregulation of the proinflammatory cytokine IL-1β (Festa et al., 2015). More recently, it was demonstrated that HIV-1 Tg rats exhibit a decrease in thin spine density within the prelimbic cortex (PrL), which negatively correlated with trials to criterion in an attentional set-shifting task, and that CXCL12 signaling rescues these deficits (Festa et al., 2020). Notably, this reduction in PrL thin spine density contrasts with the increase in striatal thin spine density observed in McLaurin et al., 2018, suggesting important brain region-specific effects of HIV-1 protein exposure on dendritic spine morphology. Collectively, these findings critically underscore the impact of chronic HIV protein exposure on mesocorticolimbic circuit function, which has important implications for drug-seeking behavior.
2.5. EcoHIV Rodent Model of HIV-1 Infection
Achieving a productive HIV infection is difficult in rodents, as they are poorly susceptible to HIV (Sawada et al., 1998; Hinkula et al., 2004). While transgenic rodent models can produce aspects of later stages of infection, such as persistent HIV protein expression, a major goal of HIV animal model development has been achieving primary viral infection in rodents that mirrors the immunological and serological milieu observed in humans. HIV infection begins with cellular endocytosis of HIV through interactions between HIV Env (i.e., gp120 and gp40) and a host cell receptor complex composed of CD4 and co-receptors CXCR4 or CCR5 (Wilen et al., 2012). While rodent cells are capable of persistent production of infectious HIV-1 (Mizrachi et al., 1992; Keppler et al., 2001), the virus does not efficiently bind to rodent cells. This is the principal challenge in producing a rodent model of sustained HIV-1 infection. Previous attempts to resolve this issue by producing transgenic rodents that express human CD4, as well as CCR5 or CXCR4, showed limited capacity to produce persistent infection in vivo (Browning et al., 1997; Sawada et al., 1998). To overcome this difficulty, a chimeric HIV-1 viral construct, EcoHIV, was created by replacing the coding region of gp120 with the envelope-coding region of gp80 from ecotropic murine leukemia virus (MLV), permitting infection and viral replication in rodents (Potash et al., 2005). Potash and colleagues demonstrated that EcoHIV produces systemic infection in mice after a single inoculation and that viral mRNA is detected in CD4+ T cells, macrophages, and microglia, which are all major target cell types of HIV-1. EcoHIV mRNA and expression of the viral proteins Tat and p24 were also detected within the brain (Potash et al., 2005). EcoHIV infection in mice also increases BBB permeability through downregulation of claudin-5, which is also observed in the gp120 Tg mouse (Jones et al., 2016). Recent evidence suggests that microinfusion of EcoHIV into the rat brain produces a detectable infection predominantly in Iba1+ cells, implicating microglia as the predominant cell type that harbors EcoHIV (Li et al., 2021b). As microglia are widely believed to be the primary HIV-1 reservoir within the CNS in humans (Wallet et al., 2019; López et al., 2021), this represents a critical translational advantage of the EcoHIV rodent model.
EcoHIV infection in the CNS induces neuroinflammation and parallel changes in neuronal morphology and synaptic function. Intracerebral infection of EcoHIV in the mouse brain induces synaptodendritic loss in the hippocampus, which is correlated with working memory impairment. This neuronal dysfunction is not associated with apoptosis (Kelschenbach et al., 2019) which mirrors findings in virally-suppressed PLWH who experience cognitive impairment with subtle neurodegeneration rather than cell loss or death (Heaton et al., 2010; Gelman, 2015). Neuronal morphology and synaptic function are also altered in EcoHIV-infected rats. For example, administration of EcoHIV into the rat cortex induces dendritic spine morphology dysfunction in NAc MSNs and medial PFC (mPFC) pyramidal neurons. Specifically, EcoHIV-infected rats show increased relative frequency of shorter dendritic spines, increased head diameter, and increased neck diameter (Li et al., 2021a, 2021b). These synaptic alterations are associated with upregulation of the expression of proinflammatory factors such as NF-κB, TNF-α, and IL-1β within the cortex (Potash et al., 2005; Li et al., 2021a). These neuronal dysfunctions in EcoHIV-rats are associated with deficits in temporal processing as well as long-term memory, which are considered aspects of HAND pathology.
One limitation of the EcoHIV model is that the virus lacks the HIV-1 Env protein gp120. As evidenced by the exogenous protein and transgenic models, HIV gp120 induces neuropathology and alters motivated behavior, including producing synaptodendritic damage. For example, intracerebral microinjection of gp120 in rats reduces spine density and causes dendritic damage in the PFC (Festa et al., 2015), and gp120 Tg mice exhibit dendritic spine deficits within the hippocampus and striatum (Bachis et al., 2016; Speidell et al., 2020). Reduced dendritic spine density within the PrL of HIV-1 Tg rats is correlated with impaired cognitive flexibility (Festa et al., 2020) and it is possible that gp120-induced synaptodendritic deficits contribute to alterations in reward circuit function. Taken together, the contribution of gp120 to HIV-related neurocognitive pathology should not be ignored. In the EcoHIV model, the potential role of gp80 within the mouse CNS has not been fully investigated. One study has reported that MLV (which contains the gp80 envelope protein) does not induce cognitive dysfunction akin to what is observed in EcoHIV-infected mice, indicating that gp80 may not model gp120-driven neuronal impairments within the CNS (Kelschenbach et al., 2019). Unlike gp120, Tat is expressed systemically by EcoHIV and can be detected within the brain (Potash et al., 2005). However, whether EcoHIV-mediated Tat expression contributes to neurocognitive dysfunction akin to other Tat models is still under investigation. These caveats should be noted when interpreting data from EcoHIV studies. Nevertheless, the evidence demonstrating that EcoHIV infects microglia/macrophages and that it induces neuroinflammation and cognitive impairment indicates EcoHIV infection in rodents is a valuable model to study HIV-associated neurocognitive disorders (Kelschenbach et al., 2019; Li et al., 2021a). Importantly, unlike HIV Tg rodent models, EcoHIV produces a sustained infection in wild type mice. However, infected mice do not appear to ever transition to an immunodeficient state after a protracted period of time in the absence of ART. Indeed, these mice show a precipitous decline in peripheral viral RNA levels by three weeks post-inoculation and exhibit anti-Gag and anti-Tat antibodies for many weeks post-inoculation (Potash et al., 2005; Gu et al., 2018), indicative of an antiviral immune response that limits the extent of EcoHIV infection. Interestingly, this is reminiscent of the asymptomatic phase of HIV infection in humans following primary infection, where individuals maintain immunocompetency. Related to SUDs, this is perhaps a unique and advantageous design feature of this model because it enables researchers to examine how persistent drug use in otherwise-healthy PLWH, under virally-suppressed conditions (which may or may not include ART), alters the course of HIV infection and subsequent changes in reward-associated cognition, behavior, and neurobiology. Nevertheless, it must be noted that once infection is established, ART treatment is ineffective at attenuating viral DNA levels in the spleen and reversing spatial memory impairments induced by EcoHIV infection (Gu et al., 2018). This represents an important caveat of the model and should be considered when designing studies that probe the impact of chronic ART treatment on neurocognitive and behavioral outcomes.
2.6. Alternative HIV-1 rodent models
Beyond these models, multiple additional rodent models of HIV have been developed in which complementary preclinical investigation would inform our understanding of co-occurring HIV-1 infection and SUD. This includes HIV-1 Nef-expressing rodent models. For example, transgenic mice expressing Nef in microglia exhibit striatal neuroimmune dysfunction, reduced levels of dopamine and DAT within the striatum, and behavioral changes such as hyperlocomotion (Acharjee et al., 2014). Another Nef transgenic mouse model, where Nef expression is induced by doxycycline treatment in CD4+ T cells, exhibits T cell activation and depletion as well as AIDS-like disease in nonlymphoid organs such as the lungs and kidneys (Rahim et al., 2009). It is possible that Nef expression alone is sufficient to induce neurobehavioral impairments, as implanting Nef-expressing primary astrocytes into the hippocampus of rats results in neuronal loss and impaired recognition memory, increased CCL2 expression, and peripheral macrophage infiltration (Chompre et al., 2013). Altogether, very few studies have identified a mechanistic role for Nef in HIV-associated neurobehavioral and cognitive impairment, and no studies to date have leveraged HIV-1 Nef models to examine the effect of Nef expression on addiction-related behavior. Future work investigating SUDs and HIV may also benefit from additional work in the severe combined immunodeficient (SCID) mouse model, where mice lack functional T and B cells and can be humanized via the engraftment of human fetal liver, thymus, and lymph node tissue (McCune et al., 1988, 1991; Namikawa et al., 1988). As reviewed in more detail elsewhere (Sil et al., 2021), other humanized mouse models, including the humanized microglia mouse model (Mathews et al., 2019) and the humanized NOD/SCID/γ chainnull (NSG) mouse model (Cai et al., 2011), may also be useful for assessing the pathophysiology and neurocognitive burden of HIV-1 infection.
3. Utilization of NeuroHIV Animal Models in Preclinical SUD Research
Co-occurring HIV and SUDs represent a unique clinical challenge. Drug use can impede successful HIV treatment outcomes and, as discussed in detail below, HIV may complicate SUD treatment efforts through modulation of drug-induced dysregulation of brain reward and motivation processes. For example, we have recently demonstrated that a candidate preclinical medication for treatment of cocaine use disorders – previously shown to successfully suppresses cocaine motivation (Powell et al., 2020) – failed to suppress cocaine relapse-like behavior in rats exposed to gp120 (Namba et al., 2023). This study highlights the need to consider comorbidities such as HIV when designing preclinical SUD models for medications development. Indeed, individuals with multiple diagnoses experience poorer treatment adherence, and integrated treatment approaches have been proven consistently superior to separate treatment of individual diagnoses (for review, see National Institute on Drug Abuse, 2020). Thus, the integration of the aforementioned preclinical HIV models with established animal models of SUDs provides an opportunity for researchers to examine how the pathophysiology of HIV, with or without chronic ART, alters the neurobiology and behavioral sequelae of addiction-related behaviors and, importantly, the efficacy of novel pharmacotherapeutics intended to treat SUDs.
3.1. Behavioral sensitization
Potentiation of behavioral responses to a stimulus after repeated exposure to that stimulus is referred to as sensitization. In the context of addictive substances, behavioral sensitization most often refers to the enhanced frequency of behavioral response to a drug following repeated exposure, which is an effect that can last chronically (Paulson et al., 1991) and occurs across a myriad of drugs (Stripling and Ellinwood, 1977; Joyce and Iversen, 1979; Robinson and Becker, 1986; Benwell and Balfour, 1992; Cunningham and Noble, 1992). Typically, behavioral sensitization within this context is observed when the same dose of a drug produces a potentiated behavioral response and/or when less drug is necessary to produce a particular magnitude of response. This phenomenon is usually measured as sensitization of drug-induced locomotion but can also include behaviors such as psychostimulant-induced stereotypy (e.g., methamphetamine-induced head twitching). Cross-sensitization, where repeated exposure to one drug can produce sensitization to another, is common between addictive drugs and points toward shared neurobiological mechanisms underlying the formation of behavioral sensitization (Vezina et al., 1989; Itzhak and Martin, 1999; Beyer et al., 2001; Cadoni et al., 2001). Importantly, many studies have demonstrated a reduction in drug-induced locomotor sensitization in rodents following treatment with FDA-approved pharmacotherapeutics used to treat SUDs (Chester et al., 2001; Häggkvist et al., 2011; Goutier et al., 2015), highlighting the value of this model towards identifying treatment targets. While the lower face validity of locomotor sensitization relative to other behavioral paradigms discussed here represents a limitation of this model, the neurobiological processes that mediate this behavior overlap with other models of drug use and relapse (Steketee and Kalivas, 2011).
Studies using intracranial microinfusions of HIV protein and transgenic rodents demonstrate that CNS exposure to HIV proteins can alter drug-induced behavioral sensitization and mesocorticolimbic neuroplasticity. One study examining the impact of CNS expression of HIV Tat on methamphetamine (METH) sensitization demonstrated that male HIV-1 Tat Tg mice exhibit enhanced locomotor sensitization and microglial activation within the dorsal striatum relative to control mice (Kesby et al., 2017). This study also demonstrated that HIV-1 Tat Tg mice have reduced expression of D1, D2, D4, and D5 dopamine receptors within the NAc, implicating altered mesolimbic dopamine transmission as a functional impairment in these mice. Similarly, male HIV-1 Tg rats exhibit enhanced sensitization of METH-induced stereotypic behavior (Liu et al., 2009) and potentiated cocaine-induced locomotor sensitization (Paris et al., 2014a). However, HIV-1 protein effects on drug-induced locomotor sensitization appear to depend on the protein exposure method, sex, and hormonal status of animal subjects. For example, intra-NAc microinfusions of Tat potentiate acute cocaine-induced locomotion but attenuate cocaine-induced locomotor sensitization in ovariectomized female rats (Harrod et al., 2008). In freely-cycling, gonadally-intact female HIV-1 Tat Tg mice, Tat induction attenuates acute cocaine-induced locomotion only during diestrus, whereas Tat induction reduces cocaine-induced locomotor sensitization regardless of cycle phase (Paris et al., 2014b). Similarly, intra-NAc Tat exposure also attenuates cocaine- (Ferris et al., 2010) and nicotine-induced locomotor sensitization in male rats (Zhu et al., 2015). In contrast to these findings, one study found that ovariectomized female HIV-1 Tg rats exhibit sensitization to cocaine-induced locomotion within the periphery of an open field arena (Moran et al., 2013). However, similar to findings from Zhu and colleagues (2015), male HIV-1 Tg rats exhibit attenuated nicotine-induced locomotor sensitization (Midde et al., 2011). While altered behavioral sensitization can implicate disrupted neurobehavioral plasticity, alone it is insufficient to model more complex motivation and reward processes that are fundamental to all SUDs. Indeed, studies examining the incentive motivational effects of the Pavlovian and operant mechanisms that underlie drug use across HIV models have revealed important insights into how HIV may alter the pathophysiology of SUDs.
3.2. Conditioned Place Preference
First developed to assess the reinforcing efficacy of opioids (Rossi and Reid, 1976; Katz and Gormezano, 1979; Mucha and Iversen, 1984), the conditioned place preference (CPP) paradigm is a common behavioral model used to quantify Pavlovian drug-context associations (for review, see McKendrick & Graziane, 2020). In preclinical addiction studies, CPP utilizes a two- or three-chamber apparatus (two primary chambers where conditioning occurs and a middle chamber), each of which consists of distinct visual, tactile, and/or olfactory cues. Testing in the CPP paradigm typically consists of three phases – the pretest, conditioning, and post-test phases. During pretesting, animals are allowed to explore the CPP apparatus to determine their baseline chamber preference. For the conditioning phase, animals receive experimenter-delivered injections of drug and are confined to one of the two primary chambers. In a biased design, drug is paired to the chamber that is opposite to the preferred chamber during pretesting. Conversely, an unbiased design randomly assigns the drug-chamber pairings to each animal. The experimenter-administered drug is repeatedly paired with the designated associated context. On alternating sessions, a neutral stimulus (e.g., saline) is paired with the opposite context, occurring either on the same day or on alternating days. After drug conditioning, animals are tested for expression of CPP, where they are allowed to explore all the entire apparatus and time spent in the drug-paired chamber is measured. Generally, increased time spent in the drug-paired chamber is associated with the rewarding efficacy of the drug. Many CPP studies using the aforementioned rodent models of HIV demonstrate that HIV may potentiate the rewarding properties of various addictive drugs, providing evidence for a role of HIV in modulating the pathophysiology of SUDs.
The majority of CPP studies in rodent models of HIV have utilized transgenic mice that conditionally or constitutively express HIV proteins. Among male HIV-1 Tat Tg mice, induction of CNS expression of Tat potentiates the expression of cocaine CPP (Zhu et al., 2022). The induction of Tat expression is also sufficient to reinstate extinguished cocaine CPP (Paris et al., 2014a; Mediouni et al., 2015). Tat induction also potentiates ethanol CPP in HIV-1 Tat Tg mice and is sufficient to reinstate this behavior following extinction training (McLaughlin et al., 2014). These findings suggest that Tat expression during reward learning and acutely during the expression of place preference is sufficient to impact reward seeking. Importantly, the effect of Tat induction on the reinstatement of drug-seeking behavior, which is a preclinical model of relapse-like behavior, indicates that Pavlovian conditioning processes that drive drug seeking in humans (O’Brien et al., 1992; Perry et al., 2014) may be impacted by HIV infection. Females within this mouse model also express potentiated cocaine CPP due to Tat induction during diestrus (Paris et al., 2014b). Akin to cocaine, male HIV-1 Tat Tg mice also exhibit potentiated morphine CPP (Gonek et al., 2018). This study found that pretreating Tat Tg mice with the CCR5 antagonist maraviroc exacerbates the potentiated morphine CPP response in these mice, which the authors hypothesized is due to complex mu opioid receptor (MOR)-chemokine receptor interactions whereby proinflammatory chemokine stimulation of CCR5 provides inhibitory tone over MOR signaling. Another recent study demonstrated that maraviroc attenuates cocaine CPP and cocaine-induced hyperlocomotion (Nayak et al., 2020). Interrogation of this effect within an HIV rodent model would shed light on whether the mechanisms that mediate HIV-induced potentiation of drug reward (e.g., CCR5 signaling) are drug-specific. Similar to HIV-1 Tat Tg mice, gp120 Tg mice that constitutively express gp120 in GFAP+ cells express greater sensitivity to the incentive motivational effects of METH, exhibiting a leftward shift in the dose response function for METH CPP (Kesby et al., 2014). In contrast to these mouse models, HIV-1 Tg rats do not show potentiation of morphine CPP but may exhibit deficits in extinction learning, although this effect appears to be sensitive to the environmental cues associated with the CPP apparatus (Chang and Connaghan, 2012; Homji et al., 2012b). Surprisingly, there are very few studies that have assessed drug-induced CPP in HIV-1 Tg rats, highlighting a critical gap in the preclinical literature. Another caveat of many of these studies is that animals are often tested for drug-induced CPP in a drug-free state making it unclear whether abstinence-dependent neuroadaptations that are modulated by HIV protein exposure drive the observed findings in the aforementioned studies. Altogether, these findings highlight significant differences in drug-conditioned reward and motivation in animal models of HIV infection. Similar to observations from drug-induced locomotor sensitization studies, factors such as drug type, sex, HIV model, and hormonal status all likely modulate the effects of HIV on drug-induced CPP.
3.3. Drug Self-Administration
Drug self-administration procedures are widely considered the “gold standard” of preclinical animal models for assessing the rewarding and reinforcing properties of psychoactive drugs and studying the neurobiology of SUDs. The key advantage of this model over others is that animals have volitional control over their drug intake, and this type of contingent drug use involves distinct neurobiological mechanisms relative to non-contingent, experimenter-delivered models of drug exposure (Namba et al., 2018). Drug self-administration employs a complex medley of operant and classical conditioning components as well as positive and/or negative reinforcement whereby animals learn that the rewarding and reinforcing effects of a particular drug are due to distinct response-outcome contingencies. These contingencies, such as the pressing of a lever for the delivery of a drug reinforcer, are associated with contextual and discrete stimuli that, through repeated pairings with the drug, acquire incentive motivational value and facilitate self-administration behavior (Estes, 1948; Davis and Smith, 1976; Arroyo et al., 1998; Carter and Tiffany, 1999; Tiffany, 1999; Caggiula et al., 2001; Perry et al., 2014). The majority of findings on the impact of HIV and its protein products on drug self-administration behavior are derived from HIV-1 Tg rodent models, except for two studies conducted in NHPs. Findings from these studies indicate significant impairments in drug-motivated behavior and associated neuroplasticity and immune dysfunction, which appear to depend on factors such as sex, reinforcer type, drug access conditions, as well as drug withdrawal and abstinence.
One of the first preclinical studies to examine the interactions between HIV infection and drug self-administration utilized the SIV-infected macaque model along with alcohol self-administration (Kumar et al., 2005). In macaques with a chronic history of alcohol consumption, both plasma and CSF viral loads remained persistently elevated beginning at 18- and 10-weeks post-inoculation, respectively, relative to control macaques with no alcohol history. Total alcohol consumption did not appear to escalate substantially post-inoculation, which may suggest that HIV infection does not affect alcohol consumption per se. However, the low sample size of this study necessitates caution when drawing such conclusions. Moreover, these animals did not self-administer alcohol daily, which is an important caveat. Related work found that SIV infection does not alter daily alcohol consumption in macaques but results in reduced levels of circulating CD4+ T cells and elevated levels of monocytes expressing CCR5 relative to control animals (Marcondes et al., 2008). Within the brain, alcohol self-administration reduced the expression of the anti-viral cytokine interferon alpha (IFNα), suggesting a reduced innate immune response to SIV infection. Together, these two studies provided early evidence that a history of drug use may alter the progression and the pathophysiological milieu of HIV. Studies building upon this work address important questions related to the impact of HIV on drug-seeking behavior.
Studies using transgenic rodent models of HIV in combination with self-administration procedures have revealed important insights into how HIV proteins dysregulate drug-seeking or -taking behavior. One study utilizing gp120 Tg mice in a two-bottle choice procedure, where mice have free access to either METH or saccharin and water within their home cage, found that Tg mice exhibit increased preference for both METH and saccharin under restricted access conditions compared to controls, but reduced METH preference under unlimited access conditions. This effect was greater in males compared to females (Kesby et al., 2014). Under certain conditions, saccharin consumption is predictive of future psychostimulant use in rodents (Gosnell et al., 1998; Perry et al., 2007), and restricted or intermittent access procedures may better represent drug use patterns observed in humans (Kawa et al., 2019). This study highlights sex and drug access conditions as critical variables when assessing the neurobehavioral underpinnings of HIV and addiction-related behaviors.
Akin to METH self-administration in gp120 Tg mice, studies suggest that HIV-1 Tg rats also exhibit enhanced sensitivity to psychostimulant reinforcement. For example, one study found that intravenous (i.v.) self-administration of cocaine in HIV-1 Tg rats produced a leftward shift in the cocaine dose response function relative to control rats under a short-access, fixed-ratio (FR) 1 schedule of reinforcement paradigm (McIntosh et al., 2015). This was accompanied by greater DAT affinity in striatal preparations from cocaine-experienced HIV-1 Tg rats versus control rats, consistent with their leftward shift in the dose response function. HIV-1 Tg rats also exhibit mPFC hyperexcitability that is augmented by abstinence from i.v. cocaine self-administration (Wayman et al., 2016). Specifically, cocaine abstinence-induced increases in mPFC pyramidal neuron excitability are further augmented in HIV-1 Tg rats, which is attenuated by inhibition of L-type Ca2+ channels. Combined with striatal dopamine dysfunction, this type of mPFC pathology could facilitate drug-seeking behavior beyond normal increases across abstinence (Tran-Nguyen et al., 1998; Grimm et al., 2001; Pickens et al., 2011). Indeed, a recent study found that following a month of abstinence from i.v. METH self-administration, HIV-1 Tg rats exhibit greater escalation of METH intake on a long-access, fixed schedule of reinforcement. Compared to wild-type rats, HIV-1 Tg rats also exhibited potentiated breakpoints in a progressive ratio (PR) task that requires animals to exert a progressively increasing effort across a test session to receive a reinforcer delivery, which is thought to test the incentive motivational value of drugs and associated cues (Richardson and Roberts, 1996; de Guglielmo et al., 2020). Interestingly, these behavioral changes were accompanied by enhanced mPFC neuroinflammation in METH self-administering HIV-1 Tg rats relative to wild-type controls with METH experience. In contrast, this group also showed no differences in long-access, i.v. oxycodone self-administration between HIV-1 Tg rats and controls after a period of forced abstinence despite cognitive deficits and transcriptomic evidence of mPFC neuroinflammation (Fu et al., 2022). Within the hippocampus, HIV-1 Tg rats, with or without a history of METH self-administration, show impairment of BBB protein expression and upregulation of MMP-9 and NF-κB expression (Ohene-Nyako et al., 2021), which is associated with enhanced drug-seeking behavior (Bozdagi et al., 2007; Russo et al., 2009; Namba et al., 2020, 2022). METH self-administering HIV-1 Tg rats also exhibit enhanced expression of ΔFosB and downstream dopamine D1 receptor expression within the NAc (Ohene-Nyako et al., 2018). Importantly, ΔFosB is a critical transcription factor that regulates many addiction-related genes, accumulates with repeated drug exposure, and promotes drug-seeking behavior (Nestler, 2008). Altogether, these studies suggest that HIV-1 Tg rats may be more sensitive to psychostimulant reinforcement and exhibit neuropathology that may promote motivation to seek drug in an abstinence-mediated manner.
It is important to note that the preponderance of this work was conducted using only male HIV-1 Tg rats, and recent drug self-administration studies in females have revealed important sex differences in HIV-induced neurobehavioral adaptations. For example, Bertrand et al (2018) demonstrated that both the reinforcing efficacy of cocaine across a range of cocaine doses and the motivation to self-administer cocaine on a PR schedule are diminished in female HIV-1 Tg rats. Furthermore, this study also showed that female HIV-1 Tg rats exhibit diminished choice for cocaine over sucrose compared to controls across a week of repeated testing. Interestingly, DAT abundance (i.e., Bmax) observed in this study was lower in Tg rats compared to controls and increased by cocaine, which parallels the cocaine-induced increase Bmax of low-affinity DAT binding sites from male HIV-1 Tg rats used in Mcintosh et al (2015). However, important procedural differences exist between these self-administration studies, such as an extensive history of reinforced lever pressing prior to cocaine self-administration in the female HIV-1 Tg rats used in Bertrand et al (2018) compared to male Tg rats naïve to such training in Mcintosh et al (2015) and Wayman et al (2016). In contrast to the latter two studies, a recent study examining cocaine self-administration behavior in male HIV-1 Tg rats using the same initial training dose as Wayman et al (2016) found that these animals are highly resistant to acquiring cocaine self-administration (Huynh et al., 2020), similar to female Tg rats in Bertrand et al (2018). This is further complicated by a recent study that demonstrated enhanced response vigor for cocaine in female HIV-1 Tg rats (McLaurin et al., 2021). Altogether, these studies highlight the need to consider sex in combination with behavioral history and task parameters when examining the neurobehavioral impact of HIV on addiction-related behaviors.
One limitation of these HIV-1 Tg rat model studies, as well as several of the aforementioned mouse model studies, is that animals are exposed to HIV proteins prior to chronic drug use, which is in distinct contrast to the SIV-infected macaque studies that demonstrated an alcohol-induced facilitation of SIV pathophysiology without SIV-induced changes in alcohol consumption (Kumar et al., 2005; Marcondes et al., 2008). One recent study showed that EcoHIV infection via retro-orbital administration in rats with a chronic history of cocaine self-administration disrupted choice behavior for cocaine versus sucrose and also impairs extinction learning (McLaurin et al., 2022). This study also showed synaptic impairments on mPFC pyramidal neurons, including a shift towards increased dendritic spine head and neck diameter and increased spine density on distal branches. This important work addresses the neurobehavioral impairments produced by HIV following a chronic history of drug use/exposure, which is a key gap in the extant literature. Considering drug use and associated risky behaviors (e.g., sexual risk taking) are associated with increased risk of HIV infection (Durvasula and Miller, 2014), it is important for preclinical models to elucidate the neurobehavioral consequences of drug history on the pathophysiology of HIV and future addiction-related behaviors following HIV infection. In a longitudinal study assessing predictors of cessation of drug use and relapse among PLWH, HIV seropositivity was associated with a shorter time to relapse following cessation (Shah et al., 2006), suggesting that abstinence-dependent neurobehavioral processes that contribute to drug relapse may be modulated by HIV. Overall, the inconsistent findings across the self-administration literature and the overall paucity of HIV-related operant self-administration studies in the extant literature highlight the need for future investigations that attempt to parse out the individual contributions of these variables to drug-motivated behavior.
4. Leveraging Advanced Behavioral Neuroscience Tools within Animal Models of Comorbid HIV and SUDs
Despite tremendous progress in our understanding of how HIV may alter addiction-related behaviors and how addictive drugs impact the pathophysiology of HIV within the CNS, there remains a need for a mechanistic circuit, cellular, and molecular understanding of the interaction between HIV and addictive drugs to gain insight into novel prevention and treatment strategies. Well-validated techniques used widely across behavioral neuroscience represent a novel future direction for preclinical HIV and SUD research. Here, we will discuss such techniques and how they have advanced our understanding of the neurobiology of SUDs. Specifically, we will briefly discuss key findings from the preclinical SUDs literature that highlight a myriad of techniques that demonstrate the cellular-, molecular-, and circuit-level contributions to addiction-related behaviors. We identify lingering questions related to the neurobehavioral sequelae of comorbid HIV and SUDs and how these techniques could help to close these research gaps. This is by no means an exhaustive review of the preclinical SUD literature employing such techniques. Nevertheless, we discuss key examples from the field of how these studies have substantially advanced our understanding of the neurobiology of SUDs and highlight how these findings may uniquely intersect with preclinical animal models of HIV.
4.1. Dissection of the Circuit-Level Contributions to HIV-Induced Dysfunction of Drug Motivation and Reward
The brain reward system function is especially vulnerable to HIV. However, many questions remain regarding neural circuit contributions to comorbid HIV and SUDs. Corticostriatal neural circuitry is particularly vulnerable to HIV infection (Illenberger et al., 2020; Nickoloff-Bybel et al., 2020), although it is not entirely clear how such circuit dysfunction modulates drug-seeking behavior. Circuit manipulation techniques such as optogenetics and chemogenetics have helped clarify the cell type- and projection-specific mechanisms underlying drug-induced dysfunction of mesocorticolimbic circuits, thus representing a novel approach towards understanding how these circuits might be differentially involved in addiction-related behaviors within an animal model of HIV.
Optogenetics refers to the manipulation of cellular activity on a millisecond timescale via optical stimulation of genetically encoded, light-sensitive proteins (Deisseroth et al., 2006; Miesenböck, 2009; Boyden, 2011). Like optogenetics, chemogenetics involves the use of genetically encoded protein receptors; however, these receptors are stimulated by small molecule ligands and are insensitive to endogenous ligands. Designer receptors exclusively activated by designer drugs (DREADDs) are the most common type of chemogenetic strategy used in preclinical addiction neuroscience studies, which are mutant muscarinic receptors that are Gq-, Gs, or Gi-coupled G-protein coupled receptors (GPCRs) that are activated by clozapine-n-oxide (CNO) (Armbruster et al., 2007; Guettier et al., 2009; Roth, 2016). More recently, a Gi-coupled kappa opioid receptor DREADD (KORD) was developed, which responds to the pharmacologically-inert compound salvinorin B (SALB), thus permitting a multiplexed DREADD approach when combined with muscarinic DREADD receptors (Vardy et al., 2015). Optogenetics and chemogenetics have been utilized across a wide range of preclinical addiction studies and are powerful tools to help reveal the underlying circuit mechanisms of addiction-related behaviors (Bernstein and Boyden, 2011; Cao et al., 2011; Ferguson and Neumaier, 2015; Vickstrom et al., 2021).
Optogenetics and chemogenetics have revealed key insights into cell type- and circuit-specific mediators of addiction-related behaviors, exemplifying an approach that will advance our understanding of how HIV dysregulates motivated behavior. Of particular relevance to the dopamine system and striatal dysregulation observed in models of HIV, optogenetic dissection of the direct and indirect striatal pathways has challenged the classic understanding of segregated D1 and D2 receptor-expressing MSN projection pathways. Canonically, it was believed that D1 MSNs project directly to output nuclei of the basal ganglia, while D2 MSNs do so through indirect innervation of pallidal neurons, with little overlap between the two pathways (Gerfen and Surmeier, 2011). However, a crucial study by Kupchik et al. (2015) demonstrated that expression of ChR2 within NAc D1 MSNs and light stimulation within the ventral pallidum (VP) results in enhanced excitatory activity in 50% of recorded VP neurons, which was also confirmed via retrograde fluorescent labeling. This study also showed that D2 MSNs can form a direct pathway that disinhibits the thalamus through inhibition of VP → thalamus inhibitory projections. Such findings provide convincing evidence that striatal MSN plasticity may have more complex and nuanced effects over mesocorticolimbic circuit function than what one might predict with the traditional direct/indirect pathway model. Other studies have revealed through optogenetics critical contributions of excitatory projections from the mPFC and basolateral amygdala (BLA) to the NAc in cue-induced motivated behavior (Stuber et al., 2011; Stefanik and Kalivas, 2013; Stefanik et al., 2016), the role of dopamine-independent glutamate transmission from the VTA to the NAc in promoting reward seeking (Zell et al., 2020), as well as the role of calcium-permeable AMPA receptors within mPFC → NAc subcircuits in cue-induced drug seeking (Lee et al., 2013; Ma et al., 2014). These studies provide a crucial foundation upon which similar studies could be conducted in HIV animal models to parse out the unique circuit contributions to drug seeking behavior within the context of comorbid HIV.
Pathway-specific chemogenetic strategies have also been used to dissect the function of specific neuronal circuits in regulating drug-seeking behavior. For example, a recent study by Yager et al (2019) utilized a Cre-lox recombinase strategy to express inhibitory DREADDs specifically within direct pathway-projecting striatal neurons to examine the role of this pathway in regulating cue-induced reinstatement of cocaine seeking. This study revealed that chemogenetic inhibition of these specific neurons suppresses cue-induced cocaine seeking only in rats screened for a “high-risk” addiction-like phenotype (characterized by higher responding for cocaine on a PR schedule and despite foot shock punishment). This group also demonstrated using a similar approach to chemogenetically tag both direct and indirect pathway-projecting MSNs that direct pathway MSNs drive, while indirect pathway MSNs inhibit, cue-induced heroin seeking only in “high-risk” rats (O’Neal et al., 2019). These studies further highlight how the underlying neural circuitry of drug seeking is contingent upon individual differences, which raises important questions regarding the neural circuit contributions to drug seeking in the context of HIV. Another recent study demonstrated that optogenetic stimulation of VTA→NAc core dopamine neurons induces reinstatement of cocaine seeking and that chemogenetic inhibition of this pathway prevents this behavior (Jing et al., 2022). As discussed previously, several studies suggest that HIV-1 Tg rats exhibit dysregulation of DAT function within the striatum, suggesting that HIV may impair dopamine transmission. Thus, understanding the role of VTA→NAc dopamine signaling within the context of HIV could reveal novel insights into the neural circuitry underlying comorbid HIV and SUDs. In addition to dopamine transmission, many studies implicate altered corticostriatal glutamate transmission as a consequence of HIV and comorbid SUDs (Potter et al., 2013; Vázquez-Santiago et al., 2014; Giacometti and Barker, 2019), and there may be dissociable effects of HIV infection on the plasticity of NAc D1 versus D2 MSNs that receive corticostriatal glutamate input to drive drug-seeking behavior (Schier et al., 2017; Barbour et al., 2021). In combination with the HIV animal models described previously, optogenetics and chemogenetics could be leveraged to isolate cell type-specific neuronal subcircuits (e.g., mPFC glutamatergic projection neurons → NAc D1 MSNs) that are responsible for HIV-induced changes in drug-motivated behavior (Pascoli et al., 2014; Garcia et al., 2018).
It remains unclear whether HIV alters the recruitment and functional influence of these circuits in cue-motivated drug seeking and relapse-like behavior. Optogenetic and/or chemogenetic dissection of circuit contributions to HIV-induced dysregulation of drug-motivated behavior would reveal crucial insight into the development of targeted medications to treat SUDs in the context of comorbid HIV. In particular, combining these approaches with viral vectors capable of transsynaptic labeling would allow for isolation of cell type-specific mesocorticolimbic subcircuits that regulate the interactions between HIV and drug-seeking behavior (Gong et al., 2007; Sjulson et al., 2016). Another outstanding question that remains is whether activity of brain reward circuitry modulates neuroimmune function and, subsequently, the pathophysiology of CNS HIV infection. Presently, it is well accepted that immune signaling crucially modulates neuronal function, particularly within the context of SUDs and associated comorbidities (Namba et al., 2021). However, the concept of neuronal signaling mechanisms (particularly those regulating associative learning processes that critically underly SUDs) shaping immune function in a reciprocal manner is a re-emerging concept that could have important implications for understanding the neurobehavioral intersections of HIV and SUDs (Ader and Cohen, 1975; Goshen and Yirmiya, 2007; Sundman and Olofsson, 2014; Dantzer, 2018; Hadamitzky et al., 2020). A recent study leveraged optogenetics to demonstrate that vagus nerve stimulation in mice confers kidney protection against ischemic injury, which is likely mediated at least in part through stimulation of cholinergic anti-inflammatory signaling (Tracey, 2007; Tanaka et al., 2021). Another recent study showed that optogenetic stimulation of phasic dopamine neuron firing within the VTA of mice stimulates serum IL-2, IL-4 and TNF-α expression, and that pharmacological suppression of VTA activity (induced naturally in males by an encounter with females) can inhibit behavior-induced increases in serum IL-2 expression (Kayama et al., 2022). This work showcases the utility of circuit-manipulating tools such as optogenetics and chemogenetics, which could be combined with preclinical HIV models to study how the activity of specific reward circuits modulate immune function and, subsequently, the pathophysiology of HIV (for reviews of this approach, see Ben-Shaanan et al., 2017; Korin & Rolls, 2018).
4.2. Cell type-specific Identification of Vulnerable Neuronal Subpopulations in Comorbid HIV and SUDs
One limitation of the classic application of optogenetics and chemogenetics is the inability to distinguish and directly manipulate distinct subpopulations of neurons that are specifically activated during discrete addiction-related behaviors. The proportion of cells that are activated by drug exposure or drug-associated experience (e.g., cue exposure) is surprisingly small – about 5% or less (Mattson et al., 2008; Koya et al., 2009; Cameron and Carelli, 2012). These cells comprise a neuronal ensemble that is specific to discrete addiction-related behaviors and is largely non-overlapping with non-drug reward ensembles (Carelli et al., 2000; Cameron and Carelli, 2012; Bobadilla et al., 2017). A variety of techniques have been used to identify and manipulate neuronal ensembles. For example, the Daun02 inactivation method allows for selective, pharmacological reduction of neuronal excitability in c-fos-lacZtransgenic rodents that produce the enzyme beta-galactosidase (β-gal) in cells that express the immediate early gene (IEG), c-Fos. In the presence of β-gal, the Daun02 prodrug is converted to daunorubicin and reduces neuronal excitability (Cruz et al., 2013, 2015). This technique was used to demonstrate that only 2–3% of NAc neurons are activated by cocaine within a cocaine-associated environment and mediate context-dependent psychomotor sensitization to cocaine (Koya et al., 2009). This cocaine context-dependent increase in Fos activity was later shown to be associated with silent synapse generation in Fos-expressing neurons of Fos-GFP transgenic mice (Whitaker et al., 2016). Another similar approach, known as Targeted Recombination in Active Populations (TRAP), utilizes transgenic mice that express a tamoxifen-dependent recombinase under the control of an IEG promotor such as c-Fos (FosTRAP mice; Guenthner et al., 2013). This permits activity-dependent expression of an effector gene, such as a fluorescent reporter, DREADDs, etc., in a cell type-specific manner. A recent study using FosTRAP mice found that peripheral immune activation activates neurons in the insular cortex, and activation of DREADDs expressed by these activated neurons is sufficient to recapitulate peripheral inflammation (Koren et al., 2021). This bidirectional relationship between CNS function and peripheral immunity, as highlighted above, represents an important area of future research into the mechanisms of comorbid HIV and SUDs. Leveraging this type of approach within an animal model of HIV would facilitate understanding of what cell types (e.g., D1 vs. D2 MSNs) contribute to drug-associated neuronal ensembles within the context of HIV infection or viral protein exposure, and whether activation of these specific ensembles alters peripheral immunity and the pathophysiology of HIV.
One limitation of these studies is that transient Fos expression is not restricted to neurons (Cruz-Mendoza et al., 2022), which can make it difficult to parse out drug-induced transcriptomic and proteomic alterations within neuronal ensembles. One study using fluorescence-activated cell sorting (FACS) to isolate neurons found that cocaine sensitization in c-Fos-lacZ transgenic rats produces a unique gene expression profile within activated neurons compared to nonactivated neurons (Guez-Barber et al., 2011). This could be potentially extended to examining Fos-activated glial cells as well and whether they participate with neurons in drug-specific ensembles. Indeed, a recent study showed chemogenetic activation of astrocytes induces robust cFos expression within hippocampal astrocytes 90 mins after CNO exposure (Adamsky et al., 2018). Recent evidence also suggests that microglial calcium events coincide with spikes in neuronal activity (Umpierre et al., 2020). Such studies highlight the potential role of glial cell activation in the formation and expression of drug memories. These experimental techniques could provide crucial insights into whether cocaine-associated neuron-glia ensembles are functionally distinct in a rodent model of HIV. Specifically, one important question that emerges from these studies is whether the transcriptomic and proteomic profiles of cocaine-associated neuronal ensembles (or neuron-glia ensembles) are altered by HIV, which could reveal novel treatment targets that are specific to comorbid HIV and SUDs.
Increases in cytosolic calcium are associated with enhanced neuronal activity (Ghosh and Greenberg, 1995; Kawamoto et al., 2012; Brini et al., 2014). Thus, genetically-encoded calcium indicators (GECIs) are an important tool for studying cell type-specific neuronal activity that could yield crucial insights into the pathophysiology of comorbid HIV and SUDs (for a thorough review of GECIs, see Lin & Schnitzer, 2016; Wang et al., 2019). Recently, a technique for observing calcium signaling and manipulating neuronal activity, known as Fast Light- and Activity-Regulated Expression (FLARE), was developed. This approach utilizes a unique transcription factor that, when activated by coincidental increases in intracellular calcium and blue light exposure, drives the expression of a transgene (e.g., a fluorescent reporter, light-sensitive opsin, etc.) that can be observed or manipulated at a later timepoint (Wang et al., 2017). This approach would be useful for tagging cellular ensembles that are activated during the acquisition, maintenance, and expression of various addiction-related behaviors and manipulating these ensembles at a later timepoint (e.g., via optogenetics). One interesting question that arises from this type of approach is whether the architecture of corticostriatal ensembles associated with drug self-administration are uniquely altered by HIV infection. The EcoHIV model would be particularly useful here, where cellular ensembles associated with drug self-administration could be tagged with a light-sensitive opsin prior to inoculation with EcoHIV. Ultimately, integration of these approaches into preclinical models of comorbid HIV and SUDs would be useful in dissecting the cell type-specific neuronal ensemble contributions to the many motivational deficits observed in rodent models of HIV.
4.3. Glial Cell Contributions to HIV-Induced Modulation in Neuronal Plasticity and Addiction-Related Behaviors
As highlighted throughout this review, glial cells are highly susceptible to HIV infection. While recent evidence suggests microglia are the primary CNS reservoir HIV that may support productive infection (Joseph et al., 2015; Wallet et al., 2019), many studies implicate astrocytes as mediators of HIV-induced neurocognitive dysfunction (Valcour et al., 2004b; Ton and Xiong, 2013). HIV and addictive drugs produce similar impairments in glial cell function within mesocorticolimbic reward circuitry (Hauser et al., 2007; Hauser and Knapp, 2014; Namba et al., 2021), thus representing a key area of interest for studying the pathophysiology of comorbid HIV and SUDs. For example, both addictive drugs and HIV proteins downregulate expression of the glial glutamate transporter EAAT-2 (i.e., GLT-1), and rescuing this deficit inhibits drug-seeking behavior (Wang et al., 2003; Knackstedt et al., 2010; LaCrosse et al., 2016; Melendez et al., 2016). Similarly, restoration of drug-induced impairments in the catalytic subunit of the cystine-glutamate antiporter (Sxc−), xCT, reduces drug-seeking behavior (Reissner et al., 2015; Namba et al., 2020). Interestingly, xCT function promotes cellular anti-HIV-1 activity in human macrophages (Rabinowitz et al., 2021). These shared glial mechanisms mediating HIV- and drug-induced plasticity may underlie the unique pathophysiological milieu of comorbid HIV and SUDs. Recent technical innovations such as optogenetic and chemogenetic control of glial cell activity, calcium imaging, and other genetic techniques to manipulate glial cell function have advanced the study of glial cell morphology and physiology within the context of substance use, thus representing a unique opportunity for future investigations to dissect glial cell contributions to comorbid HIV and SUDs.
Glial cell physiology is a key area of interest for preclinical SUD research, and recent studies utilizing optogenetics and chemogenetics to target glia have revealed important insights into non-neuronal mechanisms underlying drug-seeking behavior. One recent study utilizing optogenetics found that activation of VTA astrocytes induces real-time avoidance behavior and that genetic ablation of GLT-1 expression prevents this effect (Gomez et al., 2019). Moreover, this study also showed that concurrent optogenetic inhibition of VTA GABA neurons along with stimulation of VTA astrocytes inhibits dopamine neurons and prevents this avoidance behavior. These findings highlight how astrocytes can play a causal role in the formation of conditioned behavioral responses through modulation of neuronal microcircuit interactions. In another study utilizing a chemogenetic approach to modulate astrocyte function, Scofield et al (2015) demonstrated using a Gq-coupled DREADD driven by a GFAP promoter within the NAc core that chemogenetic activation of astrocytes promotes glutamate release and attenuates cue-induced cocaine (but not sucrose) seeking. Importantly, this effect was blocked by pharmacological inhibition of presynaptic mGlu2/3 autoreceptors, suggesting that astrocytic glutamate transmission within the NAc core contributes to presynaptic glutamate release properties and subsequent drug-seeking behavior. Morphological deficits in astrocytes (e.g., smaller cell volume and surface area) and reduced astrocyte-synapse colocalization were also observed within the NAc following cocaine self-administration and extinction. These effects were reversed by treatment with ceftriaxone, which is an antibiotic that reduces cue-induced cocaine seeking (Knackstedt et al., 2010; LaCrosse et al., 2016). One lingering question that remains from this work is whether astrocytes express spatial and/or functional plasticity around specific neuronal subtypes, such as between D1 and D2 MSNs. A recent study identified two functionally distinct forms of transient astrocyte plasticity (i.e., enhanced D2 MSN colocalization or increased extrasynaptic GLT-1 expression) that suppress cue-induced heroin seeking (Kruyer et al., 2022), suggesting that astrocytes can coordinate synaptic plasticity in a cell type-specific manner, underscoring the nuances of drug-induced astrocyte plasticity that may be relevant to HIV-induced dysregulation of astrocyte physiology.
Calcium signaling is a crucial mechanism through which astrocytes regulate neurotransmission and synaptic plasticity (Kang et al., 1998; Vesce et al., 1999; Bazargani and Attwell, 2016; Covelo and Araque, 2016). Utilizing fiber photometry and expression of the GECI GCaMP6f under control of an astrocyte-specific promoter, Corkrum and colleagues demonstrated that NAc astrocytes exhibit increases in intracellular calcium signaling in response to synaptically-released dopamine, which triggers the release of adenosine and subsequent stimulation of presynaptic autoreceptors to dampen excitatory transmission (Corkrum et al., 2020). This study reveals the critical role astrocytes play in mediating dopamine-glutamate synaptic interactions, which could have significant implications for HIV. Indeed, HIV protein exposure can impact MSN morphology and physiology and D1- versus D2-MSNs may exhibit differential vulnerabilities to HIV infection (Brailoiu et al., 2017; Schier et al., 2017; McLaurin et al., 2018).
Plasma membrane calcium ATPase (PMCA) pumps that deplete astrocytes of intracellular calcium have been used to study astrocyte calcium signaling regulation of neuronal physiology and behavior (Strehler, 2015). A recent study virally expressed a splice variant of human PMCA2 (hPMCA2w/b) and GCaMP6f in striatal astrocytes, resulting in significant reductions in both spontaneous and evoked calcium signaling (Yu et al., 2018). Reducing striatal astrocyte calcium signaling promoted repetitive self-grooming behavior and led to enhanced GABA-mediated tonic inhibition of striatal MSNs. Another study utilizing hPMCA2w/b in astrocytes found that abolishing astrocytic calcium signaling fully impairs LTP following burst firing of DA neurons and that this form of plasticity is also dependent on astrocytic expression of the D2 dopamine receptor and CB1 cannabinoid receptor (Requie et al., 2022). These findings suggest that astrocytic calcium signaling plays a critical role in modulating the enduring plasticity of VTA dopamine neurons as well as striatal MSN plasticity, which are both vulnerable to HIV-induced impairment.
Microglia are the primary cellular reservoir of HIV within the CNS and are also involved in the pathophysiology of SUDs. Thus, understanding the functional contributions of microglia to the neurobehavioral sequelae of combined HIV and SUDs is crucial. Several studies have utilized optogenetics and chemogenetics as a tool to study microglia, particularly within the context of chronic pain (Parusel et al., 2022), which represents a novel future direction for the preclinical study of HIV and SUDs. In the first study to demonstrate chemogenetic manipulation of microglia, Grace et al., 2016 demonstrated that stimulation of a Gi-DREADD within the spinal cord, the expression of which was driven by a CD68 promotor, attenuates morphine-induced pain sensitization. Chemogenetic manipulation of microglia has been leveraged to advance the study of neuropathic pain and the spinal cord across several other studies (Grace et al., 2018; Saika et al., 2020, 2021), although its use within the brain has been limited. In particular, studies manipulating brain-resident microglia within the context of learning and memory are scarce. However, one recent study using Cx3cr1-Cre transgenic mice and Cre-dependent Gq- or Gi-coupled DREADDs within the dorsal striatum found that Gq-DREADD stimulation of microglia induces conditioned place aversion and proinflammatory cytokine signaling, while Gi-coupled DREADD stimulation blocks the formation of conditioned place aversion to an inflammatory stimulus (Klawonn et al., 2021). This study also showed ex vivo chemogenetic stimulation of striatal microglia inhibits MSN excitability and that this is dependent on prostaglandin signaling from activated microglia. One gap in our understanding of the pathophysiology of comorbid HIV and SUDs is whether persistent dysregulation of microglial physiology and activation states by HIV infection directly contributes to altered functional plasticity of neurons within the reward system. The use of microglial DREADDs represents a unique approach towards closing this knowledge gap. However, one must carefully consider the HIV model used in combination with microglial DREADDs, as varying HIV models may differentially alter host immune responses and susceptibility to viral infection in ways that may impact the effectiveness of this experimental approach.
While HIV and addictive substances may share common mechanisms of action within glial cells, it is possible that they mediate these effects through distinct forms of plasticity. Examination of corticostriatal astrocyte and microglia activation and morphology within rodent models of HIV would provide critical insights into how HIV uniquely alters drug-induced glial cell plasticity. Modulators of glial cell function, such as the antioxidant N-acetylcysteine (NAC), have consistently shown success at the preclinical level at reducing drug-seeking behavior and restoring corticostriatal glutamate homeostasis (Moran et al., 2005; Reissner et al., 2015; Israel et al., 2019; Namba et al., 2020). However, clinical translation of such compounds have shown checkered success (LaRowe et al., 2013; Deepmala et al., 2015). Comorbidities such as HIV may alter the efficacy of medications targeting glutamate neurotransmission and glial cell function. Thus, studying these mechanisms within the context of HIV is paramount towards improving our understanding of the pathophysiology of SUDs.
5. Conclusions
Comorbid disorders and diseases are a norm, not an exception, for individuals living with SUDs. Rates of substance misuse among PLWH are substantially higher than among the general population, and drug use is one of several risk factors for HIV transmission. Translation of findings from preclinical studies to the clinic has faced many challenges, and many medications that have shown preclinical success in suppressing addiction-related behavior demonstrate only modest clinical efficacy. This pipeline of medications development could be improved by utilizing translational animal models that account for common comorbidities experienced by those living with SUDs. Indeed, the animal models of HIV discussed herein clearly demonstrate the unique effects of combined HIV and drugs of abuse on the brain and behavior, which raises important questions regarding the therapeutic efficacy of novel medications to treat SUDs in PLWH.
We have reviewed the utility of multiple animal models of neuroHIV and their potential for integration with preclinical SUD models. While these models have generated convergent findings across the literature with regards to the HIV-associated neuropathology they produce, each model has its own unique set of advantages and disadvantages. Likewise, there are many models of drug-seeking behavior that model different components of SUDs, such as acquisition and escalation of drug taking, tolerance and withdrawal, craving, and relapse. Combining multiple different animal models of neuroHIV with preclinical SUD models can generate complementary data sets that can address different aspects of comorbid HIV and SUD. For example, one major gap in our understanding of the biobehavioral interactions between HIV and SUDs is the characterization of immunological changes across the addiction cycle and across various addictive drugs, how HIV modulates these changes, whether the timing of infection relative to drug exposure differentially impacts these interactions, and whether these interactions depend on important variables such as biological sex, hormone status, age, co-infection status, among many others. Integration of multiple animal models can help address these gaps to reveal novel and effective treatment avenues. Moreover, complementary preclinical datasets across animal models will also help inform future clinical investigations into the neuropsychological and behavioral impact of HIV within the context of SUDs. Much of the existing clinical literature related to HAND does not directly assess important features of SUDs, such as drug craving and relapse, patterns of drug use, polypharmacy, among many others. Many of the neurocognitive domains involved in the diagnosis of HAND, including attention-information processing, learning and recall memory, and executive functioning (Antinori et al., 2007), are also implicated in SUDs, which highlights the important overlap between HAND and SUDs. However, there remains a paucity of studies examining addiction-specific neuropsychological and behavioral domains within the context of HIV irrespective of HAND. For example, brain imaging studies in humans examining neural reactivity to drug and stress cues, which can predict future drug use patterns, have revealed important insights into the neural correlates of addiction-related domains that parallel preclinical data sets (Jasinska et al., 2014; Everitt et al., 2018; Smith et al., 2023). Moreover, clinical studies have also attempted to parse the types of motivation (e.g., reward, relief, habit, etc.) underlying drug use (Grodin et al., 2019), which can be captured by complementary animal models. However, it is unknown whether HIV status is an important modulator of these neurobehavioral relationships, and preclinical animal models of HIV are a useful tool in helping close these knowledge gaps. PLWH may also experience unique environmental and psychosocial stressors that could detract from the success of standard SUD treatment efforts (Avants et al., 1998; Krishnan et al., 2018). More clinical studies are needed to further investigate this issue, and clarifying this clinical framework would facilitate preclinical studies probing neural mechanisms underlying environmental and psychosocial stress effects on addiction-related behaviors. Altogether, these caveats represent important future directions for translational bridges to be built between preclinical and clinical research that may improve SUD treatment outcomes for PLWH.
Another major limitation of the extant preclinical literature is the lack of studies investigating the impact of chronic ART on mesocorticolimbic circuit function and addiction-related behaviors. While some of the models discussed here produce neurobehavioral impairments that resemble those observed in PLWH on ART, this is not the same as investigating the effects of ART on CNS function in the presence of HIV proteins. Future studies incorporating ART into these existing models would greatly advance our current understanding of the neurobehavioral sequelae of comorbid HIV and SUDs. Furthermore, integration of novel techniques and tools in neuroscience with models of HIV will provide a pathway to (1) characterizing brain and system-wide changes in comorbid HIV and SUDs across key levels of analysis, (2) identifying potential therapeutic targets, and (3) performing preclinical testing of potential therapeutic approaches for reducing drug seeking and taking behaviors that characterize SUDs within the context of HIV. Indeed, overcoming technical limitations associated with these models and techniques while attempting to combine them in a way that is informative and translationally relevant is a challenge. Regardless, we posit that careful consideration of the strengths and limitations of each of these models and techniques prior to their combined implementation will yield unique datasets that will substantially improve our understanding of the neurobehavioral complexities underlying comorbid HIV and SUDs.